医療機器の設計においてなぜ先行品との差分に注目するのか?
ある設計変更が引き起こした教訓
2023年、ある医療機器メーカーが人工呼吸器の後継機種を開発した際、先行品から「わずか」材質を変更しただけで、市販後に予期せぬ問題が発生した。使用環境の湿度が高い条件下で、新材質が微細なクラックを生じ、気密性が低下したのである。先行品では10年以上の使用実績があり、市販後の苦情対応を通じて徹底的に改善されていた設計であったにもかかわらず、たった一つの材質変更が新たなリスクを生み出してしまった。
この事例が示すように、医療機器の設計において先行品との差分管理は、単なる形式的な手続きではなく、患者安全を守るための重要な実務プロセスである。本稿では、なぜ先行品との差分に注目しなければならないのか、その理論的背景と実務上の応用について解説する。
先行品が持つ「見えない価値」:市販後改善の蓄積
医療機器の設計において、先行品は単なる「古い製品」ではない。それは市場で実際に使用され、数多くの試練を乗り越えてきた「実証済みの設計」である。
先行品には、開発当初には予見できなかった問題に対する解決策が、設計変更という形で蓄積されている。例えば、初回承認時には想定していなかった使用環境での問題、特定の患者群での予期せぬ反応、医療従事者からの使い勝手に関する指摘など、市販後に明らかになった課題への対応が、継続的な品質改善として実施されてきたのである。
このプロセスは、ISO 14971に基づくリスクマネジメントの一環として体系的に行われる。リスクマネジメントとは、医療機器に関連するリスクを特定、分析、評価し、適切に管理するプロセスであり、製品ライフサイクル全体を通じて継続的に実施される。市販後に収集された苦情情報、有害事象報告、不具合情報は、すべてリスクマネジメントファイルに反映され、必要に応じて設計変更として結実する。
2025年現在、医薬品医療機器総合機構(PMDA)では、医療機器の市販後安全対策の強化が進められており、製造販売業者には市販後の情報収集と分析、そして必要な措置の実施がより厳格に求められている。したがって、先行品の設計には、単に「動作する」という機能的側面だけでなく、「実際の医療現場で安全に使用できる」という実証的な価値が込められているのである。
後継機種開発における先行品情報の活用
後継機種を開発する際、先行品で実施された設計変更は極めて重要なインプット情報となる。これは、先行品が市場で学んだ教訓を、新製品の要求仕様に反映させるプロセスである。
具体的には、設計開発計画の初期段階において、先行品のリスクマネジメントファイル、市販後安全管理情報、設計変更履歴を詳細にレビューする。例えば、先行品で「特定の使用条件下で誤動作が発生し、ソフトウェアのアルゴリズムを変更した」という記録があれば、後継機種ではその改善されたアルゴリズムを初期設計から組み込むことになる。
これにより、後継機種は先行品が10年かけて到達した品質レベルを、開発初期から実現できる。いわば、先行品の「進化の歴史」を継承することで、開発効率と製品品質の両立が可能になるのである。
IEC 62366-1(医用電気機器の使用適用プロセス)では、類似製品の使用上の問題から学ぶことが推奨されており、先行品の情報活用はまさにこの考え方に沿ったアプローチである。
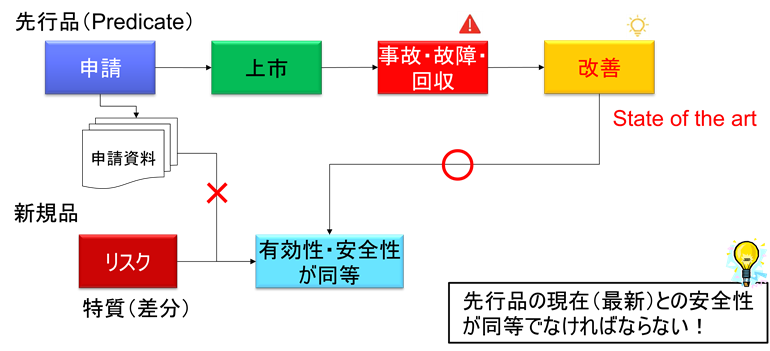
差分が生み出す新たなハザード:見過ごされがちなリスク
しかしながら、先行品との差分こそが、新たなハザードの源泉となる。ハザードとは、危害を引き起こす可能性のある潜在的な源のことであり、材質、機能、アルゴリズムなど、あらゆる設計要素の変更が新たなハザードを生み出す可能性がある。
材質変更によるハザードの例
冒頭で紹介した人工呼吸器の事例のように、材質変更は一見小さな変更に見えても、重大なリスクをもたらすことがある。別の例として、カテーテルの材質をより柔軟性の高いポリマーに変更した際、生体適合性試験では問題なかったものの、実際の使用において特定の消毒薬との反応で材質が劣化するという問題が発生したケースがある。
先行品では金属ステントが使用されており、長年の使用実績から消毒薬との相互作用は十分に理解されていた。しかし、新材質であるポリマーは、その特性が異なるため、全く新しいリスク評価が必要だったのである。
機能追加によるハザードの例
近年、医療機器のデジタル化が急速に進んでおり、2025年現在では多くの医療機器にIoT機能や遠隔モニタリング機能が追加されている。例えば、従来はスタンドアロンで動作していた血糖測定器に、スマートフォンとの連携機能を追加した場合を考えてみよう。
この機能追加により、Bluetooth通信の電磁干渉、データ送信時のセキュリティリスク、スマートフォンアプリのバグによる誤表示など、先行品では存在しなかった全く新しいハザードが生まれる。
FDAのガイダンス「Cybersecurity in Medical Devices: Quality System Considerations and Content of Premarket Submissions」(2023年更新)でも、接続機能を持つ医療機器のサイバーセキュリティリスクが詳細に論じられている。
アルゴリズム変更によるハザードの例
AI(人工知能)やML(機械学習)を活用した医療機器が増加している2025年の状況において、アルゴリズムの変更は特に注意が必要な差分である。
例えば、医用画像診断支援装置において、従来の統計的手法から深層学習ベースのアルゴリズムに変更した場合、診断精度は向上する可能性がある一方で、「ブラックボックス問題」(判断根拠の説明困難性)、学習データのバイアスによる特定患者群での性能低下、予期しない入力データへの対応不全など、新たなリスクが発生する。
FDAやPMDAでは、AI/ML医療機器に関する規制の整備が進められており、アルゴリズムの変更管理、性能モニタリング、継続的な学習への対応など、従来の医療機器とは異なる観点でのリスク管理が求められている。
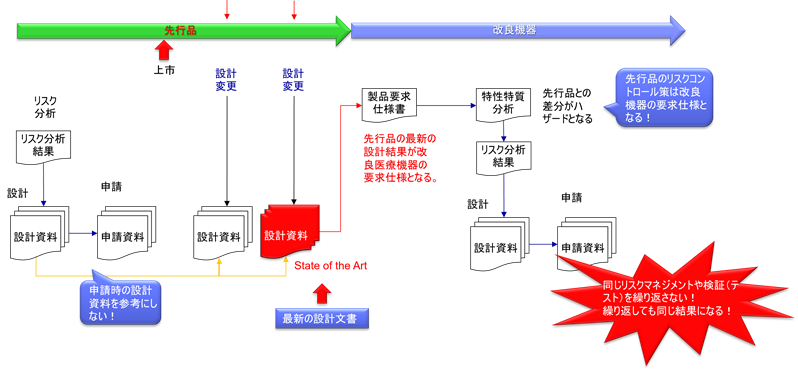
なぜ「申請時点の設計」をコピーしてはならないのか
医療機器の承認申請資料には、その時点での設計情報が詳細に記載されている。しかし、後継機種を開発する際に、単純にこの申請時点の設計をコピーすることは、重大な過誤である。
その理由は、承認後から後継機種開発時点までの間に、先行品において多数の設計変更が実施されている可能性があるからだ。これらの設計変更は、軽微変更として届出されているもの、一部変更承認申請を経ているもの、あるいは品質管理上の改善として実施されたものなど、様々な形態で存在する。
設計変更管理の実態
例えば、ある輸液ポンプの事例を考えてみよう。2015年に承認された製品が、2025年まで10年間市販されている間に、以下のような設計変更が実施されていたとする。
初年度には、特定のバイアル形状での薬液吸引時に気泡が混入しやすいという苦情があり、吸引機構の形状を変更した。3年目には、使用環境温度が高い地域での使用時にモーター性能が低下する事象が報告され、耐熱性の高いモーターに変更した。5年目には、電磁両立性(EMC)試験の国際基準が更新されたことを受け、ノイズフィルタ回路を追加した。8年目には、ユーザーインターフェースの改善要望を受け、表示画面のコントラストを向上させた。
これらの変更は、いずれも市販後の実使用経験から得られた知見に基づく重要な改善である。もし後継機種の開発者が2015年の申請書類のみを参照して設計をコピーした場合、これら10年分の改善が全く反映されず、すでに解決済みの問題を再び抱えた製品が生まれてしまう。
最新設計情報の管理体制
QMS(Quality Management System、品質マネジメントシステム)省令に基づく設計管理では、設計変更の記録と管理が要求されている。具体的には、設計履歴ファイル(Design History File, DHF)として、すべての設計変更とその根拠が文書化されている必要がある。
後継機種開発においては、先行品の最新のDHFを参照し、現時点で製造されている製品の実際の設計情報を入手することが不可欠である。これには、製造部門との密接な連携も必要となる。なぜなら、製造現場では、正式な設計変更手続きを経ずに、実質的な工程改善や部品変更が行われている場合があるからだ。
2025年現在、多くの医療機器メーカーでは、PLM(Product Lifecycle Management)システムやPDM(Product Data Management)システムを導入し、設計情報の一元管理と履歴追跡を実現している。これらのシステムにより、「現在製造している製品の正確な設計情報」へのアクセスが容易になっているが、それでもなお、関連部門との綿密なコミュニケーションは欠かせない。
米国FDAのアプローチ
FDAでは、510(k)申請において実質的同等性(Substantial Equivalence)の概念が用いられているが、差分が大きい場合や、新たなリスクを生じる場合には、実質的同等性が認められず、より厳格なPMA(Premarket Approval)が要求される可能性がある。
2025年現在、FDAではデジタルヘルス技術、AI/ML医療機器、サイバーセキュリティに関する規制の進化が続いており、これらの領域での差分管理には特別な配慮が必要である。
日本PMDAの動向
日本では、医療機器の承認審査において、同一性の考え方が重要である。後継機種が先行品と同一性を保つためには、差分が一定の範囲内に収まっている必要がある。同一性を逸脱する変更の場合、新規承認申請が必要となる。
PMDAでは、革新的医療機器の実用化促進のため、先駆け審査指定制度、条件付き早期承認制度などが運用されており、これらの制度を活用する際にも、先行品との関係性の整理が重要となる。
継続的改善の文化
医療機器の設計における先行品との差分管理は、単なる規制対応のための手続きではない。それは、過去の学びを未来に活かし、患者安全を継続的に向上させるための重要なプロセスである。
先行品が市場で学んだ教訓を尊重し、それを後継機種に活かす。同時に、差分が生み出す新たなリスクを謙虚に認識し、適切に管理する。そして、後継機種もまた市場から学び、次世代への知見を蓄積する。この継続的な改善サイクルこそが、医療機器産業における品質文化の本質である。
2025年現在、医療機器はデジタル化、AI活用、個別化医療への対応など、かつてないスピードで進化している。このような環境下では、先行品との差分はますます大きく、複雑になる傾向にある。だからこそ、差分管理の重要性は今後さらに高まっていくであろう。
開発に携わる一人ひとりが、「なぜ先行品との差分に注目するのか」という問いの本質を理解し、患者安全を最優先とする姿勢を持ち続けることが、優れた医療機器を生み出す原動力となるのである。
関連商品