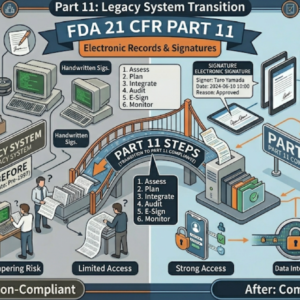
規制要件を遵守するとは
2026年最新動向アップデート(規制要件を遵守するとは)
本記事は規制要件を遵守するとはを扱ったものです。関連する規制・ガイダンスは2022〜2026年に大きく動きました。
2026年時点の主要前提
- FDA QMSR施行(2026年2月2日):21 CFR Part 820がISO 13485:2016を引用組み込み、QSITは廃止、新コンプライアンスプログラム7382.850へ。
- FDA CSA最終ガイダンス(2025年9月/2026年2月3日改訂版):リスクベース・継続的アシュアランスへ。
- ISPE GAMP 5 Second Edition(2022年7月):CSA・AI/ML・クラウド・OSS・ALCOA+を大幅追加。
- EU MDR・IVDR移行延長(Reg. (EU) 2023/607、2024/1860)とEUDAMED段階必須化。
- ISO 14971:2019+A11:2021+ISO/TR 24971:2020がリスクマネジメントの現行標準。
- FDA Section 524B(PATCH Act):医療機器サイバーセキュリティ要件。
- AI/ML:FDA最終ガイダンス「PCCP for AI-Enabled Device Software Functions」(2024年12月)。
※以下は本記事のオリジナル解説です。
筆者はこれまで多くの内部監査支援を実施してきた。
手前味噌ながら長年の経験から1回の監査で多くの指摘を出している。
その際に、各指摘についてどの規制要件に要求があるのかといった質問を受けることがある。
つまり筆者の指摘した事項は規制要件のどこにも書かれていないじゃないかといった反論である。
そうではない。規制要件は直接遵守するものではないのである。
規制要件⇒規制要件の解釈⇒手順の作成⇒実施記録の作成
となる。
規制要件はまず正しい解釈が必要である。つまり行間を読まなければならない。
各要件は何を意図しているのか、こんな風にも解釈できないかといった具合である。
規制要件の解釈は時間を追って変化することがある。
規制要件自体は変更がなくても解釈が変更になるのである。
例えば、FDAやPMDAの査察によって指摘された場合などである。
またWarning Letterなどを参考にすることもある。
では、同じ規制要件であってもなぜ各社毎に解釈を行わないとならないかについて述べよう。
それは企業毎に製造販売している製品が異なるからである。
製品が異なればリスクが異なる。
例えば、ビタミン剤や栄養剤を製造販売している企業と抗がん剤、向精神薬等を製造販売している企業では品質保証やリスク管理の程度が異なる。
また製造プロセスが異なっても解釈が異なる。
例えば滅菌工程(無菌製剤)があるかないかなどである。
FDAは2003年9月からリスクベースドアプローチといった新しい監視指導方針を示した。
各企業は自社の製品やプロセスのリスクに応じて規制要件を適切に解釈し、手順書を作成し、手順書を遵守した実施記録を作成しなければならない。