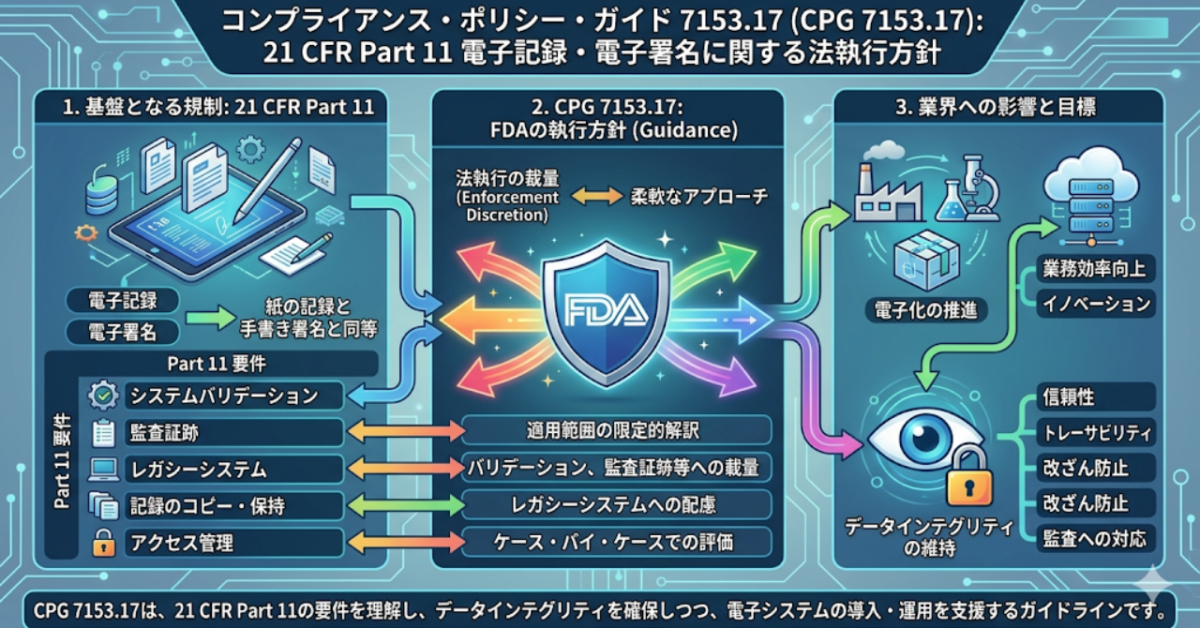
Compliance Policy Guide 7153.17とは
医薬品・医療機器業界において、FDA(米国食品医薬品局)による規制への準拠は企業活動の根幹をなす。電子記録・電子署名を規定するFDA 21 CFR Part 11(以下、Part 11)の執行に関わる文書として、Compliance Policy Guide(CPG)7153.17がある。
本稿では、CPG 7153.17の正確な位置づけと歴史的経緯を整理した上で、Part 11において特に重要とされる3つの技術的要件について解説する。
重要: CPG 7153.17は2003年2月25日にFDAにより正式に撤回(withdrawal)されている。現在の執行方針は、同年9月に発行された 「Part 11, Electronic Records; Electronic Signatures — Scope and Application」 ガイダンスに基づく。本稿はCPG 7153.17の内容と歴史的意義を解説することを目的としており、現行の規制対応には最新ガイダンスを参照されたい。
CPG 7153.17とは何か
CPG 7153.17の正式名称は 「Enforcement Policy: 21 CFR Part 11; Electronic Records; Electronic Signatures」 である。その名称が示す通り、これは執行方針(Enforcement Policy)を示したコンプライアンスポリシーガイドであり、「査察マニュアル(Inspection Guide)」とは異なるカテゴリーの文書である。
FDAの文書体系において、Compliance Policy Guide(CPG)とは、FDAスタッフに対して規制の執行戦略を助言するための文書である。具体的には、Part 11に完全準拠していない状況が発見された場合に、FDAが規制措置を追求するかどうかをケースバイケースで判断する基準を示すものだ。すなわち、「FDAがどこを見るか」ではなく、「FDAがどう判断するか」を定めた文書と理解するのが正確である。
CPG 7153.17が示していた判断基準は主に以下の4点であった。
- Part 11逸脱の性質と範囲(データの完全性を損なうか否か)
- 製品品質・データインテグリティへの影響
- 是正措置の適切性と迅速性
- コンプライアンス履歴(過去の違反歴など)
なぜ撤回されたのか
CPG 7153.17が2003年に撤回された背景には、Part 11の要件に対する解釈が過度に広範になっていたことへの懸念がある。業界からは以下の問題点が指摘されていた。
- 電子技術の使用を不必要に制限している
- 規則策定時に想定されていなかった水準のコンプライアンスコストを課している
- 公衆衛生上の利益を提供せずにイノベーションを阻害している
これを受けFDAは、CPG 7153.17を含む複数のPart 11関連ガイダンスを撤回し、Part 11をより狭く解釈する新方針へと転換した。現行ガイダンスでは、バリデーション・監査証跡・記録保持等の一部要件についてenforcement discretion(執行裁量)が認められており、すべてのpredicate rule(先行規則)の要件が引き続き執行されることが明示されている。
Part 11における3つの重要要件
CPG 7153.17は撤回済みであるが、同文書が対象としたPart 11自体は現在も有効な規則である。以下では、Part 11において特に重要とされる3つの技術的・手続き的要件を解説する。
1. 監査証跡(Audit Trail)の完全性 — §11.10(e)
監査証跡とは、システム上で行われたすべての操作——データの作成・変更・削除——を自動的に記録する機能である。Part 11 §11.10(e)は、コンピュータにより生成され、タイムスタンプが付与された監査証跡を要求しており、元のデータを不明瞭にしない形での記録保存を義務づけている。
「誰が」「いつ」「何を」変更したかが追跡可能であることが基本要件であり、これが担保されない場合、データインテグリティの観点から重大な問題とみなされる。なお、Part 11単体よりも、CGMPなどのpredicate ruleとの組み合わせで指摘されるケースが実務上は多い。
2. アクセス制御と電子署名の運用 — §11.10(d)(g)・Subpart C
Part 11は電子署名を紙の手書き署名と同等の法的効力を持つものとして認める一方で、その真正性を担保するための条件を課している。署名者の一意識別(§11.200)、署名の意図の記録(§11.50)、そしてアクセス制御の適切な実装(§11.10(d)(g))がその主要な要件だ。
実務上は、ユーザーIDとパスワードの適切な管理、退職者アカウントの速やかな無効化、共有アカウントの禁止といった運用面が問われることが多い。これらは技術的な設定だけでなく、SOPとして手続き化されているかどうかも重要な評価ポイントとなる。
3. システムバリデーションの実施と維持 — §11.10(a)
Part 11 §11.10(a)は、対象システムが意図した通りに動作することを科学的に証明する「バリデーション」を要求している。具体的には、正確性・信頼性・一貫した性能・無効または変更された記録を識別する能力の確保が求められる。
バリデーション計画書(VMP)・適格性確認(IQ/OQ/PQ)・バリデーション報告書といった一連の文書はPart 11に明示はされていないが、業界のベストプラクティス(GAMP等)として広く採用されている手法だ。システムのバージョンアップや環境変更があった際の再バリデーション実施と、変更管理記録の整備も継続的な準拠の観点から欠かせない。
まとめ
CPG 7153.17は、Part 11の執行方針を示したFDAのコンプライアンスポリシーガイドであったが、2003年に撤回されており、現在は「Scope and Application」ガイダンスが有効な指針となっている。この歴史的経緯を正しく理解することは、現行規制への適切な対応を行う上で不可欠である。
一方、CPG 7153.17が対象としたPart 11の要件——監査証跡の完全性、アクセス制御・電子署名の運用、システムバリデーションの維持管理——は現在も有効であり、その本質的な意義は変わっていない。
重要なのは、これらの要件を形式的に満たすことではなく、データの信頼性を組織の日常的な文化として根付かせることである。FDAが最終的に評価するのは、書類の整合性ではなく、現場における実質的な管理の実態である。規制の変遷を正確に把握しながら、実効性のある品質管理体制を構築していくことが、今日の規制環境を生き抜く鍵となるであろう。