
QSRからQMSRへ
2026年最新動向アップデート(QSRからQMSRへ)
本記事はFDA QSRからQMSRへの移行を解説したものです。2026年2月2日にQSRはQMSR(Quality Management System Regulation)へ移行し、21 CFR Part 820はISO 13485:2016を直接引用組み込み(incorporate by reference)する形に大きく刷新されました。
QSR→QMSR移行のサマリー
- 2024年2月2日:FDAがQMSR最終規則発行。
- 2025年12月:技術的修正(Technical Amendments)公布。
- 2026年2月2日:施行・QSIT廃止・新コンプライアンスプログラム7382.850へ移行。
- 21 CFR Part 820 = ISO 13485:2016(incorporate by reference)+ FDA固有追加要件(§820.10〜§820.45:UDI、表示、苦情、MDR等)。
FY2025 FDA Warning Letter上位指摘
CAPA(26件)、Design Controls(25件)、Complaint Files(23件)、Purchasing(15件)、Process Validation(14件)。QSR時代の旧条文番号は、QMSR下ではISO 13485:2016の対応章で読み替える必要があります。
※以下は本記事のオリジナル解説です。

QSRからQMSRへ
2022年2月23日、FDAは現行のQSR(品質システム規制:21CFR Part 820 Quality System Regulations)をISO 13485:2016に整合させる改正案を公表した。
QSRをISO 13485:2016と整合させる規制は「Quality Management System Regulations / QMSR」(品質マネジメントシステム規制)と呼ばれる。
QMSRはISO 13485:2016の要求事項を組み込むことにより、ISO 13485:2016と多くの要求を整合させる予定となっている。
その一方で、苦情ファイル等の記録の保持に関してISO 13485:2016に追加的要求事項を上乗せしている。
QSRは1996年10月7日に現行の規則が発効されて以降、20年以上に渡り米国における医療機器品質システム規制として機能してきたものである。
これまでの間、QSRは軽微な修正はあったものの、改定は一度も実施されてこなかった。
今回のQSRからQMSRの改定について、FDAには、米国における要求事項と他の規制当局が使用する品質管理システム要求事項を集約させる意図がある。
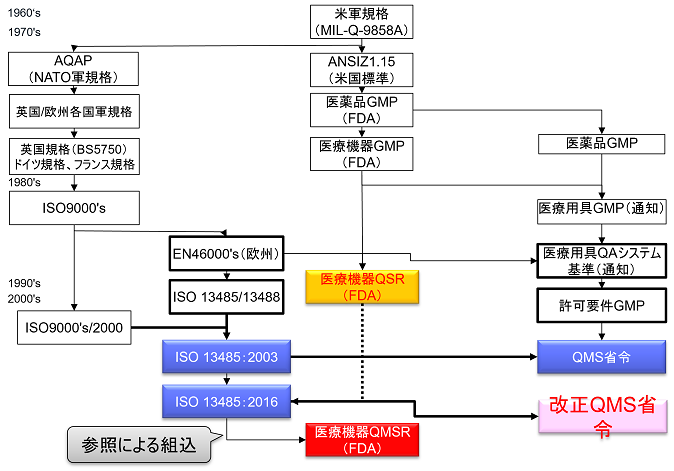
改正のポイント
FDAは、Part 820において、ISO13485:2016を参照により組み入れることによって他の規制当局が使用するQMS要求事項と整合させようとしている。
今回の改正によりPart 820の規則のタイトルがQuality System Regulations(QSR)から Quality Management System Regulations(QMSR)へ変更される。
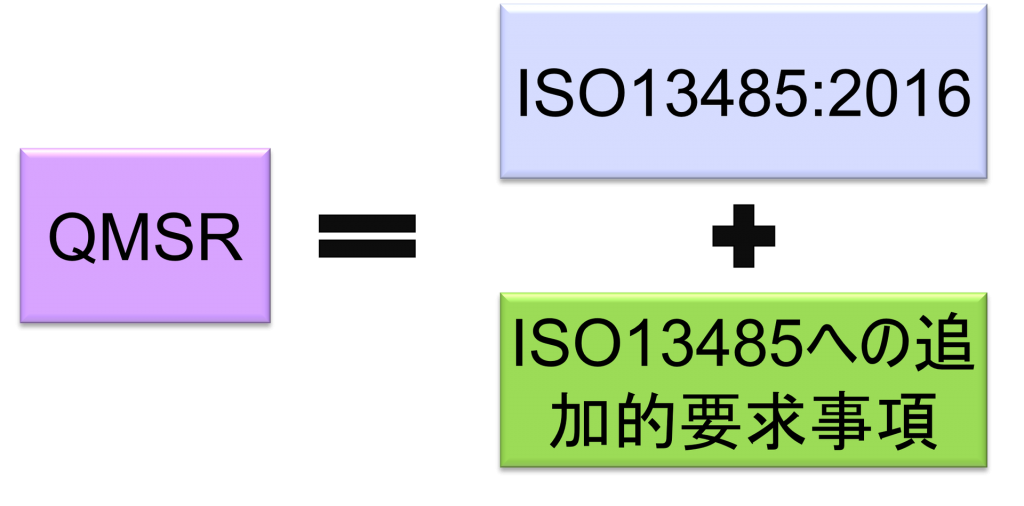
QMSRでは、原則として現行のPart 820 QSRの要求事項を取下げ、ISO13485:2016の要求事項を採用する。QMSRがISO13485:2016の要求事項自体を変更することは行っていない。
日本ではQMS省令本文(第2章)にISO13485:2016要求事項を組み入れ、第3章に日本の追加的要求事項を規定しているのに対して、QMSRではISO13485:2016を組み入れるのではなく参照の形式をとっている。
ただし、現行のQSRのうちのいくつかの定義は保持または変更を加えた上で維持される。つまりISO 13485:2016になく、QSRにある要求事項をQMSRの中で規定している。(ISO 13485:2016の要求事項はそのまま受け入れ、FDA独自の要求事項を追加する形をとっている。)
またISO 13485:2016で使用される特定の概念を明確にするためのFDA固有の要求事項および規定も追加される。
QMSRの要求事項は実質的にQSRとほぼ同じである
現行のPart 820(QSR)の要求事項は、全体としてISO 13485:2016の要求事項と実質的に同じである。したがってQMSRの要求事項は実質的にQSRとほぼ同じである。QSR特有の要求事項はQMSRにおいても維持されているためだ。
要点1.QMSRの要求事項は実質的にQSRとほぼ同じである
要点3.QMSRの遵守=ISO13485:2016の遵守に繋がるように設計されている。
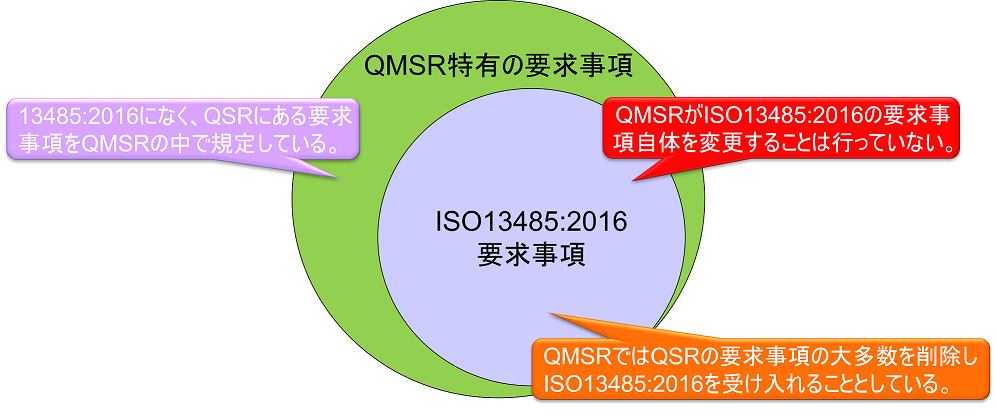
ISO13485への追加的要求事項
上図において、QMSR特有の要求事項は以下の通りである。
1.記録に関する要求事項(§820.35)
- 記録の承認者の署名および承認日の明記
- 苦情に関する記録
- 附帯サービスに関する記録
- UDIに関する記録
- コンフィデンシャルな記録の取扱い
2.ラベリングおよび包装に関する要求事項(§820.45)
3.UDI(§820.10からPart 830を参照)
4.トレーサビリティ(§820.10からPart 821を参照)
5.有害事象報告(§820.10からPart 803を参照)
6.回収(§820.10からPart 806を参照)
以下の記録の構成要素については、ISO13485の条項に従うことで達成され得るためQMSRでは規定していない
- 設計履歴ファイル(DHF)(現820.30(j))
- 機器原簿(DMR)(現820.181)
- 機器履歴簿(DHR)(現820.184)
- 品質システム記録(QSR)(現820.186) 等
ISO13485認証とQMSR採用後のFDA査察の関係
QMSRの最終化の後、品質システム査察テクニック(Quality System Inspection Technique / QSIT)の見直しが図られる。
FDA査察の受審により、ISO13485認証書が発行されることはない。
ISO13485:2016の適合証明書を有していたとしても、FDA査察は免除されない。
FDAがISO13485の認証プログラムを開発することはない。
QMSRではISO13485を取り入れるものの、企業がISO13485認証を持っていたとしても、FDAからなんらかの負担減を認められるわけではない。
関連商品












