
Part 11の4つの解けない課題
はじめに
FDA(米国食品医薬品局)が1997年3月20日に公示し、同年8月20日に発効した21 CFR Part 11は、医薬品業界における電子記録と電子署名の管理基準を定めた規制である。制定から四半世紀以上が経過した現在も、この規制には企業が直面し続ける本質的な課題が存在する。
本稿では、Part 11における「4つの解けない課題」について、実務経験に基づいて解説する。これらの課題は技術的には解決可能であっても、規制解釈の曖昧さ、経済的制約、組織的な困難さなどから、完全な解決が困難な問題として業界に認識されている。
Part 11を取り巻く規制環境2003年ガイダンスの重要性
Part 11を理解する上で欠かせないのが、2003年にFDAが発行した「Part 11, Electronic Records; Electronic Signatures — Scope and Application」というガイダンスである。
このガイダンスでFDAは、Part 11の多くの要件について「執行裁量(Enforcement Discretion)」を行使し、定型的な指摘を行わないという方針を示した。同時に、電子記録が「predicate rule(基礎となる規則)」の要件とデータ完全性を満たしているかを重視する方針に転換した。Predicate ruleとは、Part 11以前から存在するGMP、GLP、GCPなどの各種規制要件を指す。
この方針転換により、Part 11は「すべての電子データに適用される」のではなく、「predicate ruleで要求され、紙の代わりに電子形式で保管される記録」にのみ適用されることが明確になった。
最新の動向
2024年10月には、FDAが「Electronic Systems, Electronic Records, and Electronic Signatures in Clinical Investigations: Questions and Answers (Revision 1)」という最新ガイダンスを発行し、臨床試験における電子システムの取り扱いについて追加の明確化を行っている。規制環境は今も進化し続けている。
課題1:電子記録の定義 ―「タイプライターイクスキューズ」の問題
何が問題なのか
Part 11の序文には、次のような記述がある。
「Part 11は、紙による記録であって、作成後も従来の紙媒体システムで保存されるような記録の作成をする際に、単にたまたま使用されるコンピュータシステムに適用することを意図したものではない。このような場合では、コンピュータシステムは本質的には手動のタイプライタかペンのような機能をはたし、署名はいずれも従来方式の手書き署名となるものである。」
この記述が、業界で「タイプライターイクスキューズ(タイプライター言い訳)」と呼ばれる解釈論争を生み出した。

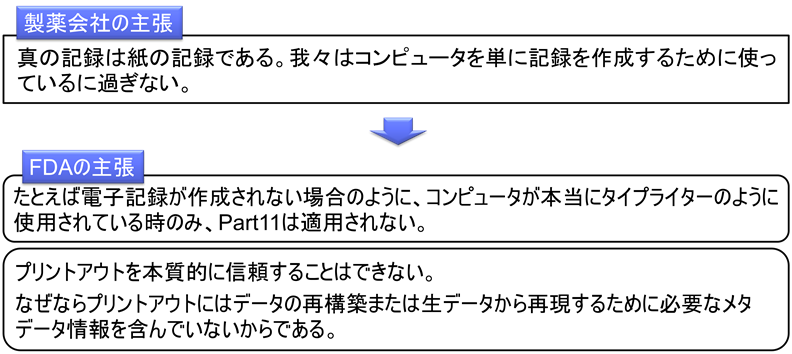
製薬会社側の主張
多くの製薬会社は、次のように主張した。
「我々はコンピュータを単に記録を作成するために使っているに過ぎない。真の記録は紙の記録である。たとえば、FDAに提出する書類をワープロソフトで作成し印刷した場合、コンピュータはタイプライターのように使用されているだけであり、Part 11は適用されないはずだ。」
この主張の背景には、Part 11の要件(監査証跡、アクセス制御、バリデーション等)を満たすことの技術的・経済的負担があった。
FDAの反論
しかし、FDAはこの解釈を認めなかった。FDAスタッフは次のように反論した。
「たとえば電子記録が作成されない場合のように、コンピュータが本当にタイプライターのように使用されている時のみ、Part 11は適用されない。プリントアウトを本質的に信頼することはできない。なぜならプリントアウトにはデータの再構築または生データから再現するために必要なメタデータ情報を含んでいないからである。」
つまり、「電子的に記録されたものは全てPart 11が適用される」という広義解釈を採用したのである。
日本での対応
日本のER/ES指針(電磁的記録・電子署名利用のための指針)では、この問題について次のように規定している。
「薬事法及び関連法令に基づいて提出する資料、原資料、その他保存が義務づけられている資料を紙媒体で作成する際に電磁的記録及び電子署名を利用する場合にあっても、可能な限り本指針に基づくことが望ましい」
つまり、日本においても「タイプライターイクスキューズ」は通用しない。ワープロで作成して印刷した場合でも、電子記録として適切に管理することが求められる。
なぜ「解けない課題」なのか
この課題が「解けない」理由は、規制解釈の曖昧さにある。どこまでが「単なるタイプライター的使用」で、どこからが「電子記録の作成」なのか、明確な境界線は存在しない。
実務的には、電子的にデータが保存される時点で、それは電子記録であると考えるべきである。しかし、企業によっては今も「紙が正(原本)である」という主張を続けているケースがあり、規制当局との見解の相違が生じ続けている。
課題2:記録と署名のリンク ―「ハイブリッドシステム」の問題
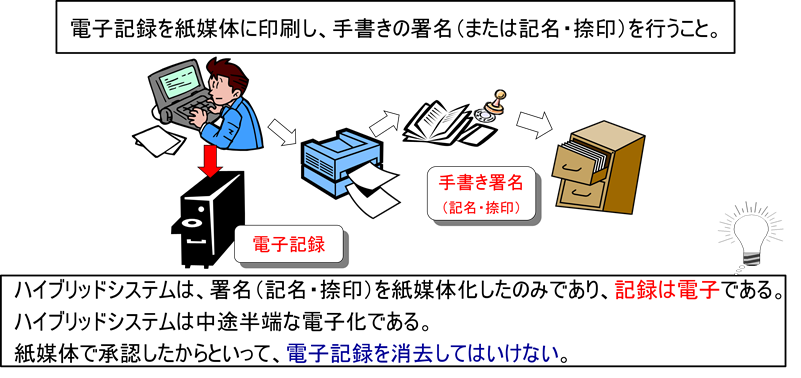
ハイブリッドシステムとは
ハイブリッドシステムとは、電子記録を紙媒体に印刷し、手書きの署名(または記名・捺印)を行う運用方法である。多くの日本企業がこの方式を採用してきた。
一見すると、これは電子化と従来の紙ベースの運用を両立させる合理的な方法に見える。しかし、規制当局の視点では深刻な問題を抱えている。
よくある主張とその問題点
企業側はこう主張する。
「当社では、責任者が記録を十分に精査して、署名(捺印)を行っている。したがって、紙が正(原本)である。」
しかし、この主張には致命的な欠陥がある。欧米の規制当局は、「多くの場合、不正(ズル)は、責任者が指示する」という前提で査察を実施する。つまり、責任者が署名したという事実は、記録の信頼性を保証しないのである。

ハイブリッドシステムの具体的問題点
ハイブリッドシステムには、以下の深刻な問題がある。
不正が容易
典型的な不正のシナリオは次の通りである。
問題のあるデータを電子的に改ざんする
改ざん後のデータを再印刷する
過去の日付(バックデート)でサインする
電子署名であればタイムスタンプが自動記録されるため、バックデートは技術的に不可能である。しかし、手書き署名では日付の改ざんを防ぐ手段がない。
「紙が正か、電子が正か」の矛盾
ハイブリッドシステムでは、「どちらが正(原本)なのか」という根本的な問題が生じる。
紙に署名しているから紙が正だと主張しても、
実際のプロセスを電子で行っている場合、規制当局は電子を正とみなす
この矛盾を解消できないまま運用を続けることになる。
電子記録の削除リスク
「紙媒体で承認したから」という理由で電子記録を削除してしまう企業があるが、これは重大な違反である。紙に署名していても、電子記録は保存しなければならない。
Excelの管理問題
ハイブリッドシステムの典型例として、Excelで入力したデータを印刷して署名するケースがある。Part 11の観点から見ると、Excelには以下の問題点がある。
- 監査証跡がとれない
- セキュリティの問題(容易に編集可能)
- 電子署名が使用できない
- バージョン間の互換性の問題
- マクロウィルス感染の危険性
- Excelを使用する場合の実務的対応
もしExcelを使用せざるを得ない場合は、以下の留意点を厳守すべきである。
入力後、すみやかに印刷し、当日の日付でサインすること(またはPDF化して電子署名を付与)
- 入力したExcelは、削除しないこと
- タイムスタンプ(ファイル日付)を変更しないこと
- セキュリティで保護された環境で管理すること(CD-R等に書き込むのが理想)
特に重要なのはタイムスタンプである。たとえば、2022年3月31日に作成されたExcelファイルに、2023年2月10日の手書き署名があったとする。これは「署名の日付よりもExcelのタイムスタンプが古い」ため、一応の整合性がある。
しかし、もし2022年3月31日の署名なのに、Excelのタイムスタンプが2023年2月10日だったら?これは明らかにデータ改ざんの疑いを示すことになる。

なぜ「解けない課題」なのか
ハイブリッドシステムが「解けない課題」である理由は、組織的・文化的な障壁にある。
日本企業では長年、「紙に押印する」ことが意思決定の証明とされてきた。完全な電子化に移行するには、業務プロセスの根本的な見直し、社内規程の改定、従業員の意識改革など、技術的対応以上の困難が伴う。
また、既存のハイブリッドシステムで数十年にわたって記録を保存してきた企業にとって、「実は電子が正だった」と認めることは、過去の記録管理の妥当性を問われることを意味する。
結果として、多くの企業が「中途半端な電子化」を続けざるを得ない状況にある。
課題3:電子記録の長期保存に関する問題保存期間の長さ
医薬品業界では、記録を長期にわたって保存する必要がある。保存期間は記録の種類により異なる。
- 製造記録:製品の有効期間+1年程度
- 治験記録:承認取得日または治験終了後3年のいずれか遅い日まで。治験依頼者が必要とする場合はさらに長期間(再審査終了まで等)
- 特定生物由来製品:製造販売業者は30年、医療機関は20年
このような長期保存には、紙の記録にはない固有の課題が存在する。
3つの深刻な問題
1.メディアを再生するためのドライブがあるか
30年前のデータを保存したメディアを想像してほしい。1990年代であれば、フロッピーディスク、MO(光磁気ディスク)、DATテープなどが使われていた。
しかし現在、これらのメディアを読み取るドライブを入手することは極めて困難である。たとえドライブが見つかっても、現代のコンピュータに接続できる保証はない。
2.当該電子記録を読み出せるソフトウェアがあるか
仮にファイルを取り出せたとしても、それを開くソフトウェアが存在するとは限らない。
具体例を挙げると、HPLCなどの分析機器で20年前に記録されたデータが、現行システムでは開けないという問題が実際に報告されている。当時使用されていた独自のファイル形式が、既にサポート終了となっているためである。
ソフトウェアの陳腐化は、技術進化の速度と保存期間要件の長さのミスマッチから生じる。20年という期間は、IT業界においては数世代の技術革新に相当する。
3.メディアへのダメージ・経年劣化
電子媒体は物理的なダメージに弱い。さらに深刻なのは、10年を超えて確実に保存できる電子媒体(CD-R、ハードディスク等)は存在しないという事実である。
CD-Rは光による読み書きを行うため、保存状態によっては5〜10年で読めなくなることがある。ハードディスクは機械的な部品を含むため、長期間使用しないと動作不良を起こす可能性がある。半導体メモリ(USB、SSD等)も、電荷の保持限界により10〜20年での劣化が懸念される。
Part 11の§11.10(c)は、保存期間中、記録が「正確かつ完全な形で取り出し可能」であることを要求している。しかし、メディアの経年劣化は避けられない物理現象であり、完全な対策は存在しない。
2つのアプローチとそれぞれの困難
長期保存の戦略として、2つの対照的なアプローチがある。
1.タイムカプセルアプローチ
電子記録が作成されたままシステムを維持する方法である。
メリット:データの完全性が保証される
デメリット:システムの陳腐化、ベンダーのサポート終了により、いずれ維持不可能になる
20年前のシステムを現在も動作させ続けることは、現実的には極めて困難である。
2.マイグレーションアプローチ
旧システムから新システムへデータを移行する方法である。
メリット:最新のシステムで管理できる
デメリット:移行プログラムのバリデーション、監査証跡を含む完全なデータ移行の技術的困難、継続的なコストの発生
特に問題なのは、元のシステムと新システムで表現方法が異なる場合、完全な移行が不可能なケースがある。その場合は、旧データ用の検索システムを別途構築する必要がある。
経済的ジレンマ
さらに、経済的な問題が状況を複雑にする。保存記録が実際に参照される頻度は極めて低い。そのため、データ移行プロジェクトは投資対効果の観点から優先順位が下がりやすい。
しかし、万が一の査察や訴訟の際に記録が取り出せないことは、企業にとって致命的なリスクとなる。この「低頻度だが高リスク」という特性が、経営判断を困難にしている。
なぜ「解けない課題」なのか
長期保存が「解けない課題」である理由は、技術進化の速度と保存期間の長さが根本的にミスマッチしているためである。
20年後、30年後の技術環境を予測することは不可能であり、現在の「最善策」が将来も有効である保証はない。結果として、企業は「継続的にコストをかけて対応し続ける」しかない。これは「解決」ではなく「対処の継続」である。
課題4:レガシーシステムの対応
レガシーシステムとは
Part 11制定(1997年)以前から稼働している古いシステムを「レガシーシステム」と呼ぶ。これらのシステムは、当然ながらPart 11の要件を前提として設計されていない。
しかし、これらのシステムは現在も医薬品業界の重要な業務を担っている。製造実行システム(MES)、品質管理システム(QMS)、研究室情報管理システム(LIMS)など、基幹業務を支えるシステムの多くがレガシーシステムである。
FDAがレガシーシステムを免責しない理由
2003年のガイダンスで、FDAはレガシーシステムについて執行裁量を行使すると述べた。しかし、これは「免責」を意味するものではない。
FDAがレガシーシステムを免責しない理由として、以下が挙げられている。
1.FDAが電子データシステムへアクセスして査察を行うことを拒否される可能性がある
レガシーシステムは、外部アクセス機能が不十分な場合が多い。これにより、FDAの査察官が電子記録を直接確認できない状況が生じる。
規制当局にとって、電子記録を直接確認できないことは、データの完全性を検証できないことを意味する。紙に印刷されたものだけを見せられても、元のデータが改ざんされていないかは確認できない。
2.システムに対して許可されないアクセスを許してしまう
レガシーシステムは、現代的なアクセス制御機能(役割ベースのアクセス制御、多要素認証等)を持たない場合が多い。
IDやパスワードの再利用を許してしまうことも問題である。たとえば、退職した従業員のIDがそのまま残っていたり、複数の人が同じIDを共有していたりするケースがある。これでは、「誰が」記録を作成・修正したのか特定できない。
3.システムがバリデーションされない状態になることを許してしまう
レガシーシステムは、導入時にはバリデーションされていても、その後の修正・変更が適切に管理されていない場合がある。
変更管理(Change Control)が不十分なため、「今このシステムがどのような状態なのか」が不明確になる。ドキュメントが散逸し、バリデーション状態が確認できない状況も珍しくない。
4.電子記録が多くの方法で改ざんされ、また発見できない状態を許してしまう
レガシーシステムの最大の問題は、監査証跡機能が存在しないか、不完全であることである。
監査証跡がなければ、データの改ざんを検知することは不可能である。誰が、いつ、何を変更したかの記録がなければ、不正があっても発見できない。
また、データベースに直接アクセスできる管理者がいる場合、その操作が記録されないこともある。システム管理者による不正を防止・検知する手段がないのである。
なぜ「解けない課題」なのか
レガシーシステム問題の根本的な難しさは、「動いているシステムを止められない」というジレンマにある。
技術的困難
レガシーシステムを置き換えるには、以下の困難がある。
他システム(ERP、PLM、QMS等)との複雑な統合を再構築する必要がある
長年の運用で蓄積された「暗黙知」の継承が困難
新システムでの運用方法の確立とユーザートレーニング
経済的困難
システム刷新には数億円から数十億円の投資が必要
投資回収の見通しが立ちにくい(生産性向上効果が測定困難)
データ移行プロジェクトの優先順位が低くなりがち
組織的困難
医薬品製造は24時間365日稼働しており、長期間の製造停止は収益に直結
システム変更は製造販売承認の変更申請が必要な場合がある
関連部門(IT、品質保証、製造、規制等)の調整が複雑
これらの理由により、レガシーシステムは「解けない課題」として存在し続けている。













