第2章FDAの医療機器規制の歴史
2.1 連邦食品・医薬品・化粧品法起源
アメリカは、19世紀後半から20世紀初頭にかけて、ヨーロッパからの粗悪な医薬品や食品による重大な健康被害に遭っていた。人々が、品質の悪い食品や医薬品によって命を落としていたという事実である。
1800年代、アメリカはイギリスの民地から独立した国家として、公衆衛生における重大な課題に耐えしていた。 当時の市場には、虚偽の表示や危険な成分を含む製品が横行し、消費者このような状況を根本的に改善するために、1938年に連邦食品・医薬品・化粧品法(FD&C法)が制定された。
FDAの取り始めの主な目的は、ミスレベリング(パッケージに記載された内容と実際の内容が異なること)を厳しくしたのだ。具体的には、以下の4つの主要な要件を定めた:
- 食品は食べて安全であり、衛生的な条件で製造されること
- 医薬品と医療機器はその目的の用途に対して安全かつ有効であること
- 化粧品は安全であり、正しい成分から作られること
- 表示と梱包は真実な情報を考慮したものであり、虚偽の情報を含まないこと
アメリカの行政制度の特徴として、司法・立法・行政の三権が完全に独立しており、行政府が法律案や改正案を直接提出することはできない。考えた場合は、議員に許可されなければならない。FDAがFD&C法のもとに告発した事件は裁判所がその審判を行うこととなる。
連邦法は州国際取引の際に適用され、州をまたがない取引の場合には州法が適用される。 CFRの規則などは法律ではないため、刑事訴追はできないという特徴も持っていた。そうした法的な取り組みの中で、FDAは消費者の安全を守るための新たな規制体系を構築していくものである。

2.2 FDA医療機器規制の歴史
1978年7月21日、FDAは連邦官報(43 FR 31508)で最終規則を発行し、FD&C法セクション520(f)に基づく医療機器のCGMP要求事項を確定した。 18日に発効し、その一部1978年7月21日に公表されたGMP規則は医療機器の製造および品質管理に対する要件を定めているが、1990年に「医療機器安全法」(SMDA)が成立するまではほとんど変更がなかった。
1978年に公表されたGMP規則は、医療機器の製造および品質管理に対する要件を定めていたが、医療機器分野における規制はまだ発展途上にあった。ただでなく、設計上の利点が安全性に大きく影響することが徐々に認識されるようになってしまいました。
2.3 品質システム規則:医療機器の製造に関する基準(医療機器GMP)
SMDAはGMP規則に設計管理(設計) FDAは、SMDAのもとにGMP規則を改正して新たな設計管理規定を加え、同時にGMP規則をできる限り国際基準(ISO 9001)に含まれる品質システムの要件に適合する方針をとった。
2.4 FDA医療機器規制の歴史
1990年に制定された医療機器安全法(Safe Medical Device Act:SMDA)は、医療機器規制における歴史的な転換点となった。が製品設計の欠陥に発見しているという重大な発見でした。
それまでの規制は主に製造プロセスに焦点を当てていたが、SMDAは設計管理の重要性を明確に認識した。 FDAは、医療機器のCGMP規制に設計管理を追加する重要な許可を与えられた。
具体的な変更点として、医療機器に関するGMP規則から「GMP」という名称を外し、「クオリティシステム規制(Quality System Regulatory)」へと変更しました。この名称変更は、品質管理が一貫した製造プロセスを超えたものです。含むようなアプローチであることを象徴的に示していた。
法律は、設計管理の要件を盛り込むことを認め、製造行為に設計を含めることを明確にした。医療機器の設計段階での品質管理が、製品の安全性と有効性を確保する上で限り重要であることが広く認識されるようになった。かつ系的なアプローチを提供し、製造業者に対してより高さで総合的な品質管理を求めるものとなった。
2.5 品質システム規則:医療機器の製造に関する基準(医療機器GMP)
1996年10月7日、食品医薬品局(FDA)は、21 CFR Part 820として医療機器の品質システム規則(Quality System Regulatory, QSR)を公開しました。この規則は医療機器の設計、購入、製造、包装、表示、保管、設置およびサービスにおける品質管理の基準を確立するものであった。
規則のタイトルは「品質システム規則:医療機器の製造に関する基準(医療機器GMP)」と定められ、医療機器産業における品質管理の新たな標準を示した。を含めて使用適合性を満たすための機器の能力を支える特徴と特質の総体を意味していた。
2.6 Code of Federal Regulation (CFR) Title 21 Part 820
QSRは標題を「品質システム規則:QSR(品質システム規則)」とし、その適用範囲を人体用に意図された全ての医療用完成機器の設計・購買・製造・梱包・ラベリング・保管・取り付けおよび付帯サービス構成はサブパートAからOまでの15セクションに分かれており、主な要件として以下を含んでいる。
対策および予防措置
品質システム要求事項
設計管理
文書管理
識別およびトレーサビリティ
生産および工程管理

2.7 21 CFR Part 820 QSR(目次)


2.8 品質システム規則:医療機器の製造に関する基準(医療機器GMP)
品質システム規則の一般規定は、以下の重要な点を定めている:
各製造業者は設計または製造される特定の医療機器に対して正しいな、そしてこの規則の要件に適合する品質システムを確立し、維持しなければなりません。
・各規則に定める要件は、最終機器が安全かつ有効であり、またFDC法令遵守を保証する意図をもつ。
・規則は最終機器の構成物または部品の製造業者には適用されないが、このような製造業者に対してガイダンスとしてこの規則の規定を設けることをお勧めする。
・QSRの適用対象は、一部の例外を除く全クラスの医療機器である。 QSRは、クラスⅠ機器にも適用されるが、わずかに例外がある。器、弾性包帯は、QSR勧告クラスⅠ機器である。設計管理は、一部の例外を除き、クラスⅠ機器には適用されない。例外として、コンピュータソフトウェア自動化クラスⅠ機器などがある。
2.9 Food and Drug Administration Modernization Act: FDAMA
FDA近代化法
1997年、FDAの行政改革を目的としてFD&C法を改正する「FDA近代化法」がアメリカ議会を通過し、同年11月21日、クリントン大統領の意思によって施行された。機器に関する規制の強化と緩和が中心となって、多くの規則の改正や制定、あるいはガイダンスの作成を要求し、これまでにない広範な改革を求めるものであった。要求する規則やガイダンスの仕事のほとんどを完了させた。
特に注目される改革には、小児研究の推進、ファストトラック計画による承認審査の迅速化、医薬品の適応外使用(表示外適応)、FDA規則の改善、医療機器の規制の強化と緩和、食品の規制包装材料の事前承認を廃止などが含まれている。これらの改革は、医療機器産業に大きな影響を与え、より効率的で効果的な規制の限界を確立することに貢献した。
2.10 FDA医療機器規制の歴史
2022年2月、QSRをISO 13485:2016に統合させるPart 820の改正案が公表された。これは1996年のQSRの制定以来、初めての大きな改正となる。FDAの規制をISO規格と調和させることは、製造業者にとって大きな猶予がある。現在は米国および他国で医療機器を販売する製造業者の多くがFDAとISO双方の要求事項に準拠する必要があるが、QMSRが施行された場合、製造業者は調和された一連の要求事項に準拠すれば良いことになる。
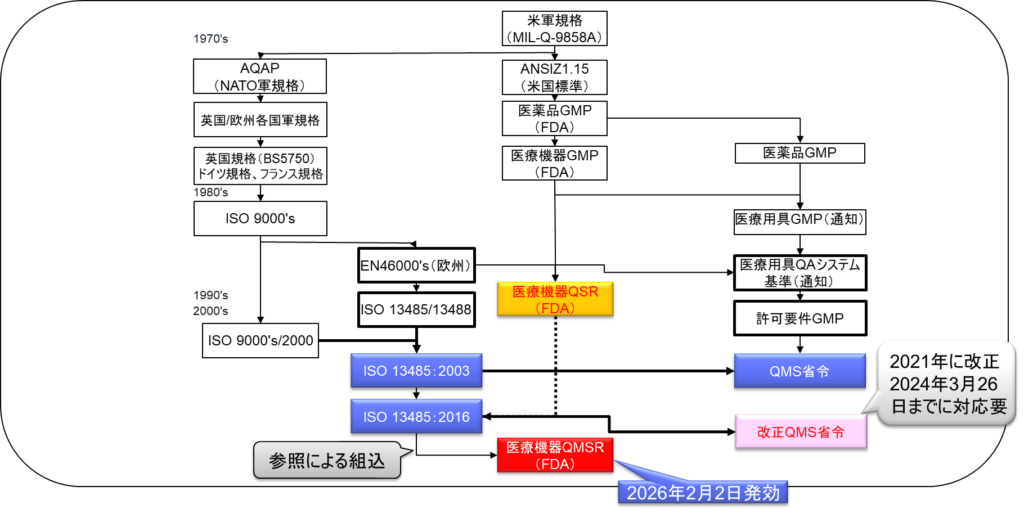
2.11 医療機器品質マネジメントシステム規格の歴史の変遷
医療機器の品質管理システム規格は、1960年代から現在に至るまで以下のような発展を遂げてきた:
1960年代には米軍規格(MIL-Q-9858A)から始まり、1970年代にはAQAP(NATO軍規格)、英国/欧州各国軍規格が策定された。確立され、1990年代にはEN46000’s(欧州)、ISO 13485/13488、医療機器QSR(FDA)が整備されました。
2000年代に入ると、ISO 13485:2003が制定され、さらに2016年にはISO 13485:2016へと進化しました。日本では2021年にQMS手続きが修正され、2024年3月26日までの対応が求められますそして米国では医療機器QMSR(FDA)が2026年2月2日に発効することが決定された。
この歴史の変遷は、グローバルな調和の流れを反映しており、特に最近は国際的な整合性を重視する方向にある。と供給を確保しながら、製造業者の規制対応の負担を軽減することを目指している。
この歴史的展開は、医療機器産業における品質の重要性が増大し、グローバル化に伴う国際的な調和の必要性が前向きに管理されていることを示している。これにより、医療機器製造業者は複数の規制要件への対応にパフォーマンスを低減しつつ、高い品質基準を維持することが可能となることが期待されている。
米国FDAは、このような国際的な規制調和の流れの中で重要な役割を果たしており、QMSRの見直しを大切にし、より効率的で効果的な規制の安定の安定を目指している。の品質と安全性を確保しつつ、グローバルな医療機器市場における協議な取引を促進することにも貢献するものと考えられている。の向上に大きく注目することが期待されている。