
QSITとは何か
QSITの基本概念
QSITとは「Quality System Inspection Technique(品質システム査察技法)」の略称であり、米国食品医薬品局(FDA)が2000年1月から医療機器分野の査察に採用してきた手法である。1998年に開発が開始され、1999年8月に最終化されたこの手法は、医療機器の品質システム規制(QSR, 21 CFR Part 820)への適合性を評価するために開発された。その目的は、製造者が製品の安全性および有効性を確保するための品質システムを適切に構築・運用しているかを確認する点にある。
QSITの構造と特徴
QSITは実際には7つのサブシステムから構成されているが、査察では主に4つの主要サブシステムに焦点を当てる。
主要サブシステム(4つ)
- 管理責任(Management Controls)
- 設計管理(Design Controls)
- 是正・予防措置(CAPA: Corrective and Preventive Actions)
- 製造および工程管理(Production and Process Controls)
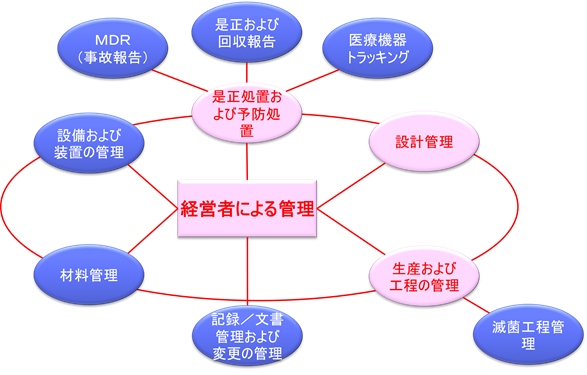
サブシステム(3つ)
- 記録、文書、変更管理
- 設備・施設管理
- 材料管理
QSITは単なるチェックリストに基づく監査とは異なり、品質システムの本質的な有効性を検証することに主眼を置いている。システマティックかつ深掘り型の査察により、表面的な適合だけでなく、実際の運用がシステム設計どおりに機能しているかを評価する点が大きな特徴である。
QSITの廃止発表:2025年4月の歴史的転換点
2025年4月に開催されたMedCon 2025において、FDAは四半世紀にわたり使用されてきたQSITを廃止することを正式に発表した。この発表により、QSITは2026年2月2日のQuality Management System Regulation(QMSR)施行と同時に完全に廃止されることが明確となった。
重要な点として、FDAは「QSIT No. 2」や「QSIT 2.0」といったスタンドアローンの後継文書を作成しないことも明らかにした。新しい査察プロセスは、医療機器コンプライアンスプログラムの中で説明される予定である。
QMSRへの移行:国際標準との調和
QSITの廃止は、より大きな規制改革の一環として行われる。FDAは2024年2月にQMSR最終規則を発表し、従来のQSRを置き換える新規制が2026年2月2日から完全施行される。この新規制の主な特徴は
- ISO 13485:2016との整合性の確保
- リスクベースアプローチの強化
- MDSAP(Medical Device Single Audit Program)監査アプローチとの調和
- より柔軟でプロセス指向の査察方法論の採用
トップダウン型査察手法の継承と発展
QSITの核心であった「トップダウンアプローチ」は、新たな査察体系においても基本的に継承される。これは、品質システム全体の構造やポリシー、マネジメントレビューの体制といった上位概念から着手し、下位の手続きや記録へと順次掘り下げていく手法である。
新アプローチでは、査察官が企業の規模、製品のリスクレベル、過去の遵守履歴などに応じて、査察の深度や範囲をより柔軟に調整できるようになる。これにより、リソースの効率的な活用と、リスクの高い領域への集中的な査察が可能となる。
移行期における企業の対応
2026年2月2日の完全移行まで、医療機器メーカーは以下の準備を進める必要がある。
- ISO 13485:2016への適合性確認:既存の品質システムのギャップ分析
- リスクマネジメントの強化:ISO 14971に基づくリスク管理プロセスの見直し
- 文書化要求事項の更新:QMSRの新要求事項に対応した文書体系の構築
- 内部監査プログラムの改訂:新しい査察アプローチを反映した監査手法の導入
- 教育訓練の実施:全従業員への新規制要求事項の周知徹底
まとめ:新時代への準備
25年間にわたり医療機器査察の基準として機能してきたQSITの廃止は、単なる査察手法の変更以上の意味を持つ。これは、医療機器規制のグローバルハーモナイゼーションへの大きな一歩であり、企業にとっては品質システムを国際標準に準拠させる重要な機会となる。
トップダウン型の査察哲学は維持されながらも、より柔軟でリスクベース、そしてプロセス指向のアプローチが採用されることで、医療機器の安全性と品質の継続的向上がさらに促進されることが期待される。2026年2月の完全移行に向けて、企業は早急に準備を開始し、この歴史的な転換期を成功裏に乗り越える必要がある。
関連商品












