電子生データの取り扱い
電子生データの取り扱い
製薬業界において、電子生データ(Electronic Raw Data)は、製造や試験、研究の過程で最初に生成されるデジタル形式のデータを指す。例えば、実験室の分析機器から直接収集される測定結果や、製造管理システム(LIMS:Laboratory Information Management System)に入力されたプロセスデータなどが、その代表的な例である。
電子生データには、紙の記録とは大きく異なる特徴がある。最も重要な特徴は、複製を作成した場合でも、各コピーが全て同等の生データとしての価値を持つという点である。このため、どの時点で取得したデータを電子生データとして扱うかを、あらかじめ手順書等で明確に定義しておくことが極めて重要となる。一般的には、測定機器やシステムにおける最初の記録時点のデータを電子生データとして定義することが多い。
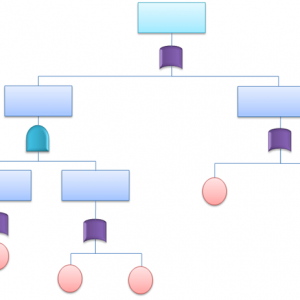
【EDCを利用した際の電子生データの取り扱い例】
時系列的に「電子症例報告書原本」をプロトコール(またはSOP)で定義しておくこと
1.ASP利用中:ASPサーバ上の電子記録
2.ASP終了後:CD-Rに焼かれた電子記録(pdf)
3.社内保管後:EDMSに登録した電子記録
4.EDCの改訂時:移行後の電子記録
5.災害発生時:上記電子記録の正式なバックアップ
データの信頼性確保
電子生データの信頼性を確保するため、規制要件では「ALCOA原則」という基本的な考え方が採用されている。ALCOAとは、Attributable(帰属性:誰が作成したかが明確である)、Legible(判読性:内容を明確に読み取ることができる)、Contemporaneous(同時性:データを生成した時点で記録されている)、Original(原本性:最初に記録された形式を保持している)、Accurate(正確性:誤りのない正確な記録である)の頭文字を取ったものである。
これらの原則を満たすため、電子生データを扱うシステムには、データの改ざんや不適切な消去を防ぐための機能が必要となる。特に重要なのが監査証跡(Audit Trail)と呼ばれる機能で、これによってデータへのあらゆる操作の記録が自動的に保存される。
また、システム自体の信頼性も重要である。GxP(Good Practice)と呼ばれる品質基準に従って、システムが適切に動作することを検証(バリデーション)し、継続的にその性能を維持する必要がある。さらに、データへのアクセスを適切に管理するため、ユーザーの権限設定や認証機能も不可欠である。
紙の記録との違い
紙の記録と電子生データでは、その取り扱い方に大きな違いがある。紙の記録の場合、原本の複写(コピー)を正式な記録として使用する際には、「True Copy」(認証済み複写)という手続きが必要となる。これには、原本との同一性確認、True Copyである旨の明記、作成者の署名と日付の記載、そして必要に応じて確認者による署名が求められる。
一方、電子生データの場合、システムによる完全な複製が可能であり、適切なアクセス管理と監査証跡が維持されていれば、全ての複製が同等の真正性を持つ。ただし、長期保存に関しては、システムの陳腐化対策など、独自の課題が存在する。
データの修正に関しても大きな違いがある。紙の記録では、修正内容、修正者の署名、日付、修正理由を手書きで記載し、元の記録が判読可能な状態を保つ必要がある。これに対し電子生データでは、システムが自動的に変更履歴を記録し、アクセスログも含めた詳細な監査証跡が残される。
規制要件への対応
電子生データの取り扱いに関しては、世界各国の規制当局が様々な要件を定めている。米国FDA(食品医薬品局)の21 CFR Part 11は、電子記録と電子署名の使用要件を規定しており、システムのバリデーションや監査証跡の維持を求めている。欧州連合のGMP Annex 11は、電子記録管理に関する包括的な要件を示している。また、PIC/S(医薬品査察協同スキーム)のガイドラインは、より詳細なデータインテグリティの要件を規定している。
まとめ
電子生データの管理には、紙の記録とは異なる特有の課題がある。しかし、適切な管理体制を整備し、システムのバリデーションを実施することで、むしろ紙の記録よりも高い信頼性を確保することが可能となる。重要なのは、データの発生時点を明確に定義し、その後の管理方法を確立することである。これにより、製薬プロセスの品質保証と、規制要件への適合を同時に達成することができる。
製薬業界におけるデータの信頼性確保は、最終的には患者の安全性に直結する重要な課題である。電子生データと紙の記録、それぞれの特性を理解し、適切な管理戦略を構築することが、高品質な医薬品の製造と供給を支える基盤となる。