
なぜFDAはDHFの作成を要求するのか
なぜFDAはDHFの作成を要求するのか
DHFに関しては、先月も解説したが、誤解が多いので再度取り上げてみたい。
DHF(Design History File)は、ISO 13485:2016において設計・開発ファイルと呼ばれるものに相当する。
しかしながら、そもそも DHFの場所や構成について、FDAによる要求事項はない。
DHFに関する良くある間違い
筆者が長年に渡ってコンサルティングを実施する中で、しばしばDHFに関する以下のような誤解を経験する。
- DHFは規制当局の査察官に見せるために作成する
- DHFを作成していなければ指摘を受ける
- DHFは、設計開発文書の改訂履歴を管理するものである
- 各設計文書(技術文書)の最新版のみを登録する
- 各設計文書(技術文書)を個別に登録する
- 各設計文書(技術文書)毎の版数(履歴)を管理する
- 各設計文書(技術文書)のドラフト版も登録する
- 物理的にすべての文書をバインダーに綴じる
- 必ず製品毎に作成しなければならない
これらはすべて間違いである。
フォルダーに設計開発文書が集まっていれば良い
DHFは、個々の文書の履歴を管理するものではなく、設計開発の履歴(ステージ毎に作成した文書)を管理するものである。
ステージ毎(例:設計インプットステージ、設計アウトプットステージ)にフォルダーを作成しておき、設計文書を保存しておけば良いのである。
DHFは、設計の各ステージにおいて、成果物を該当フォルダー内に集めることで作成すること
DHFは個々の製品(ファミリー)毎に作成しなければならず、格納すべきファイルも製品毎に異なる
なぜFDAはDHFの作成を要求するのか
では、なぜFDAはDHFの作成を要求するのだろうか。
それは以下の理由からによる。(デザインコントロールガイド参照)
- 設計履歴を管理するため
第一にDHFに望まれることは、製造業者や規制当局が散逸する設計履歴文書に対して、必要な時に必要な情報にアクセスしやすくすることである。
設計関連文書は、各部門のバインダーに綴じられていることが多い。例えば、品質システム計画はQA部門、設計開発はチーフエンジニアといった具合だ。
さらに、個人のパソコン、メモ、電子メールなど色んな場所に設計の履歴が散逸していては困る。 - 苦情(故障、修理を含む)が発生した場合、適切な設計文書(製造仕様書を含む)をもって調査出来るようにするため。
製造日における設計文書・製造仕様書の塊が即座に取り出せるようにしておかなければならない。例えば、5年前、3年前、先月では、設計仕様や製造仕様が異なるのである。
DHFは、製品(ファミリー)毎の時系列における、ある時点での最新の設計文書(技術文書)のまとまりである。
つまり、ある時点で、スナップショット写真を撮るイメージである。けっして、個々の設計文書別にファイリングするものではないことに注意が必要である。 - 設計開発が、設計計画書に基づいて実施されたことを証明するため。設計開発文書が紛失することを防止するため。
全ての設計履歴文書は、製造業者の所有物であり、社員や委託先のものではない。 - 以下のポイントを改善するため。
研究室にあるノートは私物ではなく、企業の資産である。
エンジニアの積極的なプロジェクトヘの参加の結果、別々のノートが各プロジェクトで維持され、エンジニアリング司書に提出される。
研究室のノートは、もし社員が会社を辞めるときは引き渡される。
製品開発管理者は、社員の研究室ノートをレビュし、定期的に記録が完全で、正確で、読みやすいことを確認する。
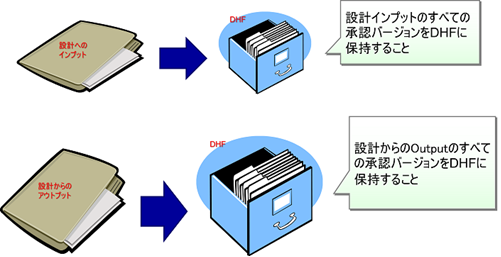
DHFはインデックス(目録)である
DHFは物理的にバインダーに綴じる必要はなく、各設計文書(技術文書)の所在をインデックス(目録)として管理すれば良い。
設計文書が電子フィルの場合は、ハイパーリンクで参照できれば良いし、紙媒体の場合には、書庫やキャビネットの場所など保存場所(参照先)を記録しておけば良い。
必要な設計文書が速やかに探し出せることが重要だからだ。
DHFに登録するタイミング
DHFには、設計管理に関するあらゆる文書を、設計審査(DR)で承認されたまとまりとして保存する。 設計審査(DR)の開催のタイミングを検討する必要がある。
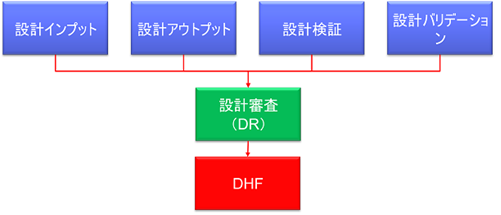
FDAが要求するDHFに管理するべき文書とは
FDAは21 CFR 820.30において、以下の3種類の記録をDHFに保存することを要求している。
820.30(e)設計審査(DR)
820.30(f)設計検証
820.30(g)設計バリデーション
おおよそ以下の文書がDHFに登録されることになる。
設計のタスクと成果物を明確にする詳細な設計開発計画書
承認された設計インプットと、設計アウトプット
設計審査の文書
バリデーション文書
適切な場合、管理された設計文書と変更管理記録
つまり、計画書で定義されている設計に関するすべての記録はDHFに登録されている必要がある。 設計のInput、Output、Design Review、Verification、Validationの各時点での成果物などである。
関連商品










