世界一わかりやすいMDRセミナー【第15講】 適合性評価手順

製造業者は機器のMDRの適合を行う場合、適合性評価手順(Conformity Assessment Procedures)に基づく必要がある。
製造業者は、医療機器のリスクに応じた「適合性評価手順」を見極める必要がある。
適合性評価手順はArticle 52 「適合性評価手順」に記載されている。
「製造業者は、機器を上市する前に、当該機器の適合性評価を、Annex Ⅸ~Ⅺ に規定されている該当する適合性評価手順に従って実施しなければならない。」
適合性評価手順は機器のクラス等により異なる。
- 日本、米国では、行政当局(または権限を委託された外部機関)が新しい医療機器について市販前の承認(認証)を与えるというシステムを採用 l製造業者の観点から見ると、医療機器の市販前に審査を受け、その結果として「承認(認証)を与えられる」形となる(受動形)
- 欧州では製造業者が自ら規制要求事項への適合を「宣言」し、CEマーキングを表示するという考え方を採用(能動形)
機器のクラス分類がClass Is,Ir,ImまたはClass IIa以上の場合、製造業者が適合宣言をするためのプロセスの中でNBの適合性評価が必要
そのため、製造業者が完全に自らの意思のみで宣言できるわけではない。
しかしながら、最後は自ら宣言をして、製造業者の法的責任の下でCEマーキングを表示するのが欧州の考え方である。
そのため、CEマーキングは「取得する」という表現は使用しない。 「添付する」または「表示する」を使用する。
MDDと異なった適合性評価手順は以下のとおりである。
- Class IIb埋め込み医療機器
- クラスⅢとほぼ同じ扱いとなる。
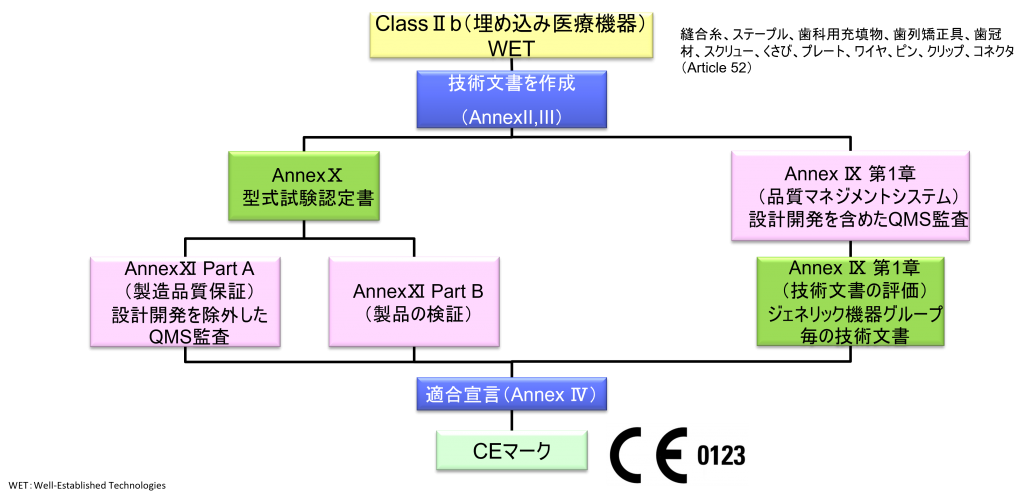
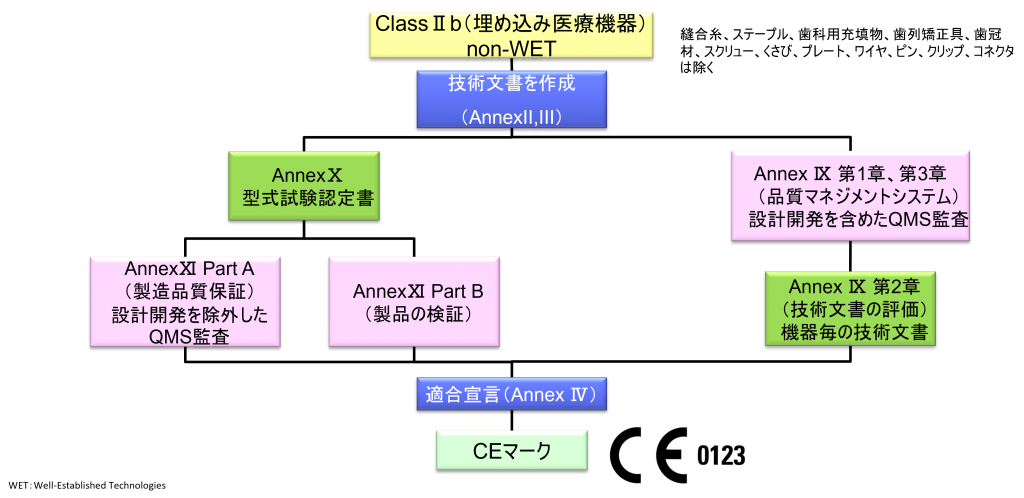
- ただし、縫合糸、ステープル、歯科用充填物、歯列矯正具、歯冠材、スクリュー、くさび、プレート、ワイヤ、ピン、クリップ、コネクタは除く
- ClassⅠ再使用可能な外科用器具
- クラスⅠ滅菌医療機器、測定機能を持つ医療機器とほぼ同じ扱いとなる
機器により、以下の特別な追加適合性手順がある。
- 医薬品を含む医療機器の場合、Annex Ⅸ Section 5.2またはAnnex X Section 6も適用すること。
所轄官庁(CA)または欧州医薬品庁(EMA)の評価がある。 - ヒト血液由来を含む機器は、出荷前にバッチ検証が必要。
- ヒト組織または細胞由来を含む医療機器の場合、Annex Ⅸ Section 5.3またはAnnex X Section 6も適用すること。
所轄官庁(CA)の評価がある。 - 人体開口部を経てまたは皮膚に適用され、人体に入れられ、人体内で吸収または局所的に分散されることを意図した物質、または物質のコンビネーション医療機器は、Annex Ⅸ Section 5.4 またはAnnex X Section 6も適用すること。
所轄官庁(CA)または欧州医薬品庁(EMA)の評価がある。
適合性評価手順(Article 52)
- 製造業者は、機器を上市する前に、当該機器の適合性評価を、Annex Ⅸ~Ⅺに規定されている該当する適合性評価手順に従って実施しなければならない。
- 製造業者は、上市されていない機器の使用を開始する前に、当該機器の適合性評価を、Annex Ⅸ~Ⅺに規定されている該当する適合性評価手順に従って実施しなければならない。
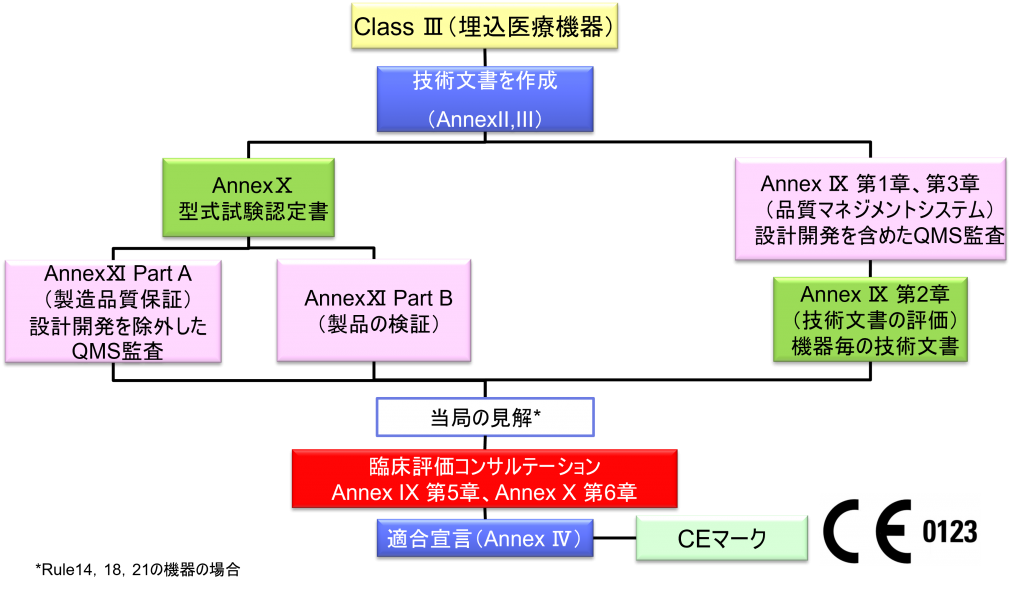
- クラスⅢの機器(ただし、カスタムメイド機器または治験機器は除く)の製造業者は、Annex Ⅸに定められているように、適合性評価の対象とならなければならない。あるいは、当該製造業者は、Annex Xに定められている適合性評価とAnnex Ⅺに定められている適合性評価を併せて適用することを選択してもよい。
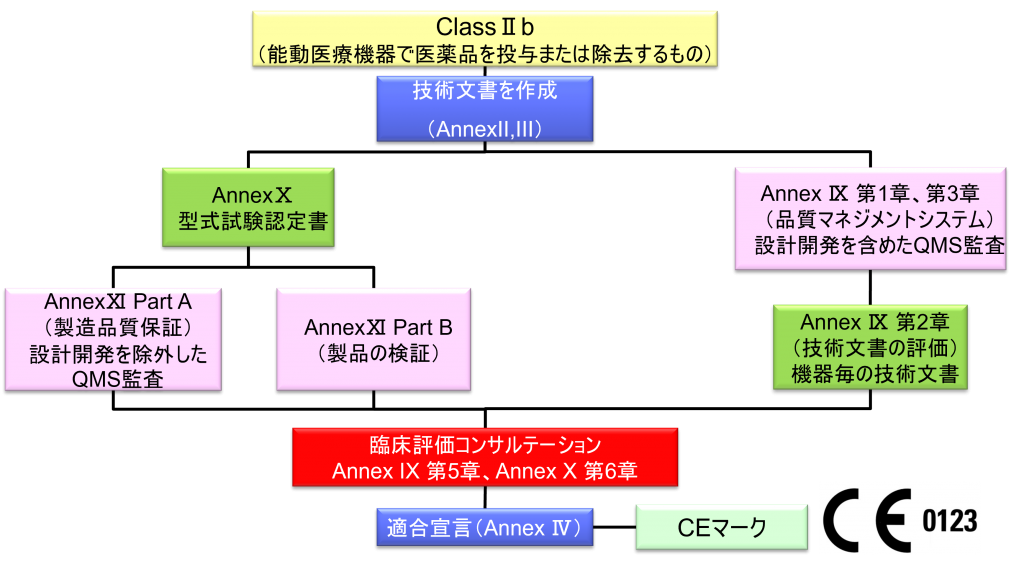
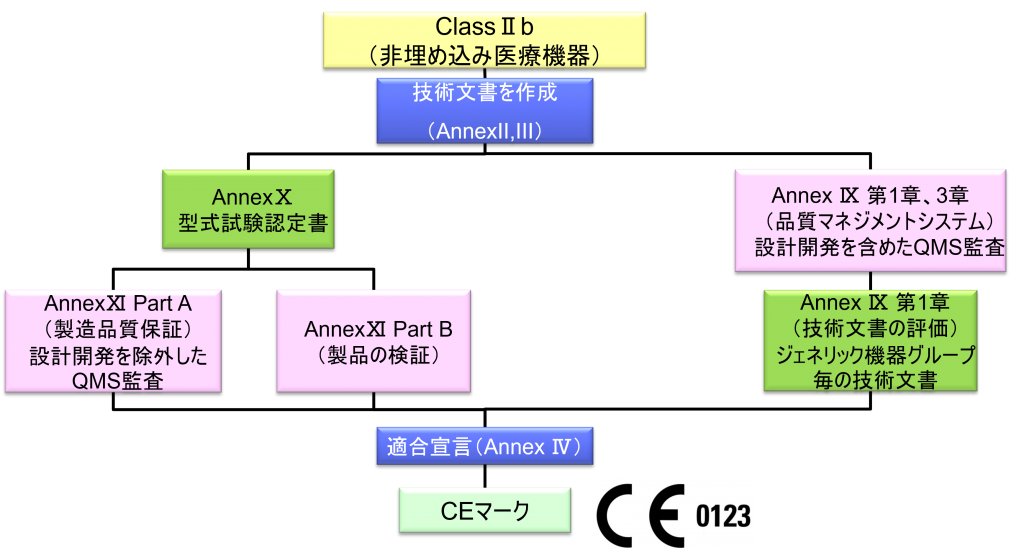
- クラスⅡb機器(ただし、カスタムメイド機器または治験機器は除く)の製造業者は、Annex Ⅸの第I章および第Ⅲ章に定められているように、適合性評価の対象とならなければならない。これには、ジェネリック機器グループごとに少なくとも一つの代表機器に関する、当該Annexの第4節に定められている技術文書の評価を含める。ただし、縫合糸、ステープル、歯科用充填剤、歯列矯正具、歯冠、ねじ、くさび、プレート、ワイヤ、ピン、クリップおよびコネクタを除くクラスⅡb埋込機器については、Annex Ⅸの第4節に定められているように、技術文書の評価は、全ての機器に適用しなければならない。あるいは、当該製造業者は、Annex Xに定められている型式審査に基づく適合性評価とAnnex Ⅺに定められている製品適合性検証に基づく適合性評価を併せて適用すること選択してもよい。
- 本条項の第4項の中段に挙げた免除機器に使用されている、確立した技術に類似した技術がその他のクラスⅡb埋込機器に使用される正当性が認められた場合、または患者、使用者などの健康と安全若しくはその他の公衆衛生面を保護する正当性が認められた場合、欧州委員会は、その他のクラスⅡb埋込機器のリストヘの追加、または機器のリストからの削除といった修正を行うために、Article 115に従って委任法令を採択する権限が与えられる。
- クラスⅡa機器(ただし、カスタムメイド機器または治験機器は除く)の製造業者は、Annex Ⅸの第I章および第Ⅲ章に定められているように、適合性評価の対象とならなければならない。これには、機器のカテゴリーごとに少なくとも一つの代表機器に関する、当該Annex第4節に定められている技術文書の評価を含める。あるいは、当該製造業者は、Annex Ⅺの第10節または第18節に定められている適合性評価と併せて、AnnexⅡおよびⅢに規定されている技術文書を作成することを選択してもよい。技術文書の評価は、機器のカテゴリーごとに少なくとも一つの代表機器に対して適用しなければならない。
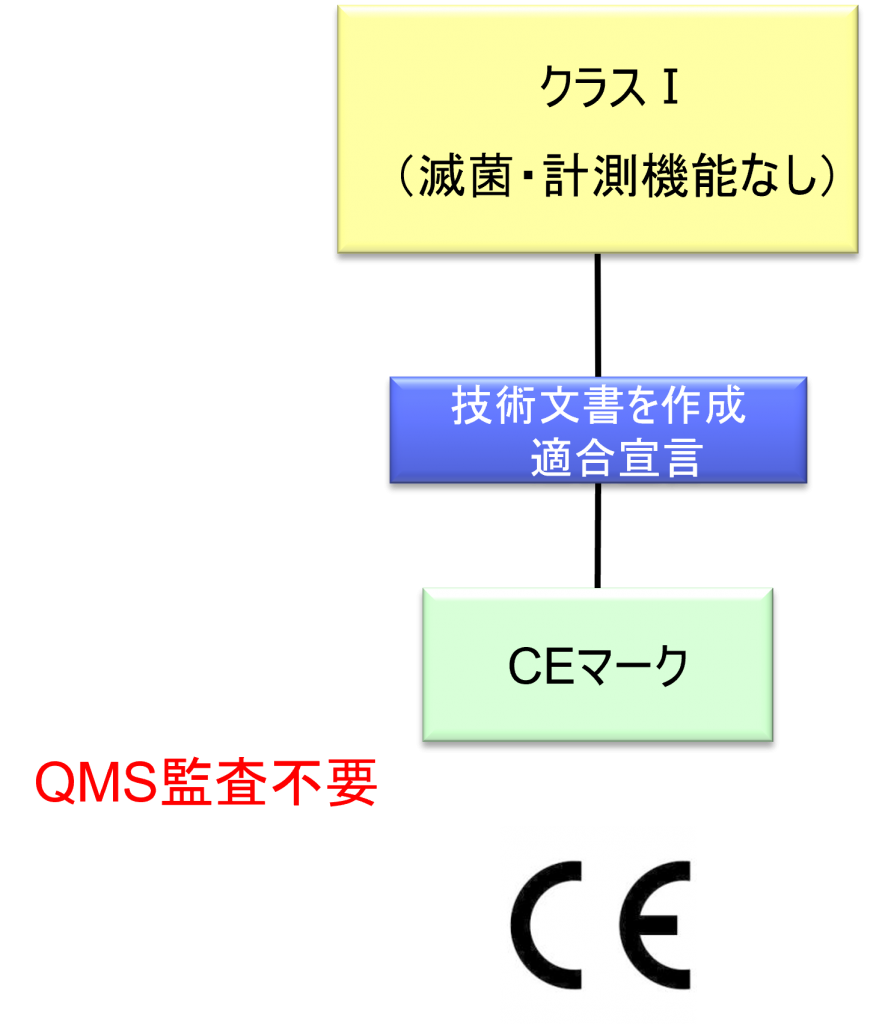
- クラスI機器(ただし、カスタムメイド機器または治験機器は除く)の製造業者は、AnnexⅡおよびⅢに規定されている技術文書の作成後、Article 19に規定されているEU適合宣言書を発行して製品の適合性を宣言しなければならない。それらの機器が滅菌状態で上市される、計測機能を有する、または再使用可能な外科用器具である場合、製造業者は、Annex Ⅸの第I章および第Ⅲ章またはAnnex ⅪのパートAに規定されている手順を適用しなければならない。ただし、次の事項については、本手順におけるNBの関与は制限されなければならない。
a.滅菌状態で上市される機器の場合、滅菌条件の設定、確保および維持に関連する事項
b.計測機能を有する機器の場合、計量要件への適合性に関する事項
c.再使用可能な外科用器具の場合、機器の再使用、特に洗浄、消毒、滅菌、保守および機能検査、並びに該当する取扱説明書に関する事項 - カスタムメイド機器の製造業者は、Annex XIIIに規定されている手順に従い、そうした機器を上市する前に、当該Annexの第1節に規定されているステートメントを作成しなければならない。前段に準拠して適用される手順に加え、クラスⅢカスタムメイド埋込機器の製造業者は、Annex Ⅸの第1章に定められている適合性評価手順の対象とならなければならない。あるいは、当該製造業者はAnnex ⅪのパートAに定められている適合性評価の適用を選択してもよい。
- Article 1第8項の前段に規定されている機器の場合、本条項の第3、4、6または7項に準拠して適用される手順に加え、適用対象となる場合は、Annex Ⅸの第5.2節またはAnnex Xの第6節に規定する手順も適用しなければならない。
- Article 1第6項の(f)または(g)およびArticle 1第10項の前段に従って、本規制の対象である機器の場合、本条項の第3、4、6または7項に準拠して適用される手順に加え、適用対象となる場合は、Annex Ⅸの第5.3節またはAnnex Xの第6節に規定する手順も適用しなければならない。
- 人体開口部を介して人体へ入れる、または皮膚に塗布することが意図され、人体に吸収される、または局所的に分散される物質若しくは物質の組み合わせから成る機器の場合、第3、4、6または7項に準拠して適用される手順に加え、適用対象となる場合は、Annex Ⅸの第5.4節またはAnnex Xの第6節に定められている手順も適用しなければならない。
- NBが設立されている加盟国は、第1項~第7項および第9項~第11項に規定されている手順に関する全ての文書または特定の文書(技術文書、監査、評価および検査の報告書を含む)が、加盟国が決定した欧州連合の公用語版で入手できるよう要求してもよい。そのような要求事項を定めない場合、それらの文書は、NBの同意する欧州連合の公用語で提供されなければならない。
- 治験機器は、Article 62~Article 81に規定されている要求事項の対象としなければならない。
- 欧州委員会は、NBによる適合性評価手順の統一的な適用を保証することを目的として、実施法令により次の点に関する詳細な取り決めおよび手順を規定してもよい。
a.クラスⅡaおよびクラスⅡbの機器については、Annex Ⅸの第2.3節第3項および第3.5 節に、クラスⅡaの機器については、Annex Ⅺの第10.2節に規定されている代表機器の技術文書の評価の頻度およびサンプリング
b.機器のリスクのクラスおよび種類を考慮し、Annex Ⅸの第3.4節に従ってNBが実施する予告なしの現地監査および抜取試験の最低頻度
c.Annex Ⅸの第3.4節および第4.3節、Annex Xの第3節、並びにAnnex Ⅺの第15節に従った抜取試験、技術文書の評価および型式審査においてNBが実施する物理的試験、臨床試験またはその他の試験
前段に規定された当該実施法令は、Article 114第3項に規定されている審査手順に従って採択されなければならない。
クラスⅠ機器(滅菌・計測機能なし)の適合性評価ルート
クラスI*(滅菌医療機器、計測機能付き医療機器、再使用可能な外科用器具)機器の適合性評価ルート

Class Ⅱa

ClassⅡb (能動医療機器で医薬品を投与または除去するもの)
ClassⅡb(非埋め込み医療機器)
ClassⅡb(埋め込み医療機器)WET(Well-established Technology)
ClassⅡb(埋め込み医療機器)non-WET
Class Ⅲ非埋込医療機器の適合性評価ルート

Class Ⅲ埋込医療機器の適合性評価ルート
お役立ち動画

世界一わかりやすいMDRセミナー
【第1講】欧州制度概要
【第2講】MDR概要
【第3講】規則実行に関連する機関等
【第4講】移行スケジュール
【第5講】MDRの要点
【第6講】用語の定義
【第7講】概要製造業者の責務
【第8講】規制遵守責任者
【第9講】一般的なMDR対応の流れ
【第10講】MDRが適用される機器・製品
【第11講】クラス分類
【第12講】安全性および性能の要求事項
【第13講】技術文書
【第14講】臨床評価
【第15講】 適合性評価手順
【第16講】欧州指定代理人
【第17講】輸入業者
【第18講】販売業者
【第19講】UDI
【第20講】EUDAMEDへの登録














