
なぜ3ロット必要か
なぜ3ロット必要か
FDAが1987年に発出した「Guideline on General Principles of Process Validation」は、医薬品の製造プロセスにおけるバリデーションの基本的な原則を定義しており、プロセスバリデーションの実施に「連続した3ロット以上」が要求されている。このガイドラインにおいて、3ロットの検証が必要とされる理由は以下の通りである。
1987年ガイドラインのポイント
1. 統計的な信頼性の確保
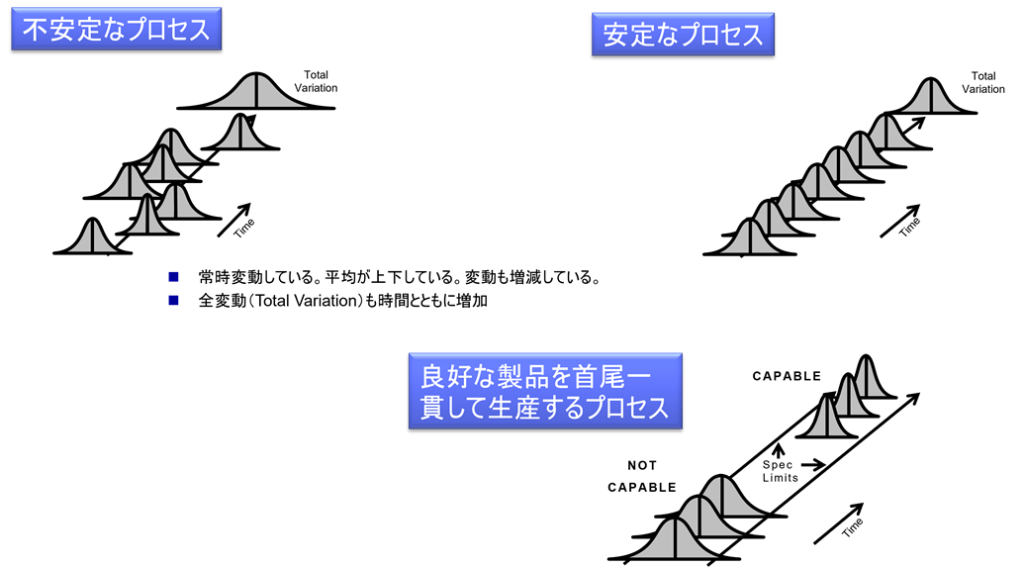
3ロットの検証は、プロセスが安定しているかどうかの判断に最低限必要な数とされている。3回の連続した実施により、安定した結果が得られれば、プロセスの一貫性と再現性がある程度確認できるため、バリデーションの信頼性が増す。
2. 再現性の確認
1ロットや2ロットでは偶然に良好な結果が得られた可能性があり、プロセスの性能を正しく評価できない場合がある。2ロットのデータでは、必ず直線が引けてしまうため、そのプロセスが本当に安定しているかどうかの判断が難しいという点もある。3ロット目を追加することで、データが直線状に並んでいるか、すなわちプロセスが一貫性を持つかどうかをより正確に評価することが可能となる。これにより、プロセスの安定性と再現性をより確実に確認することができる。
3. プロセスの変動を捉える
製造プロセスには多くの要因が関与しており、それらの要因が製品の品質に与える影響を評価するためには、十分なロット数での検証が必要である。3ロットは、少なくとも製造時に生じる変動や偏差を捉え、それに耐え得るプロセスであることを評価するのに十分とされている。
4. 品質保証の観点
製薬業界では、製品の品質が患者の健康に直接影響するため、高いレベルの品質保証が求められる。3ロットの検証は、顧客や規制当局に対してプロセスが確立され、安定した高品質な製品を提供できることを示す基準とされている。
5. プロセスバリデーションの定義
このガイドラインでは、プロセスバリデーションを「特定のプロセスが、あらかじめ定めた仕様や品質特性に適合した製品を一貫して生産できることを、高度に保証する証拠を確立すること」と定義している。
6. 文書化の重要性
バリデーションは文書化された証拠を基に行われるため、適切な文書管理が不可欠である。文書がなければ、プロセスの信頼性を保証することはできない。
7. プロセスの安定性
製造プロセスは、温度、湿度、原材料の質などの変動要因に対して安定している必要がある。これにより、製品の品質を常に維持することが求められる。
8. PPQ(Process Performance Qualification)
FDAは、プロセスの性能を確認するために、少なくとも3ロットの製造を通じてデータを収集することを推奨している。この手法は、プロセスが安定していることを示すための重要なステップである。
2011年ガイドラインの概要
FDAが2011年に発出した「Guidance for Industry Process Validation: General Principles and Practices」は、医薬品の製造プロセスが一貫して高品質な製品を生産できることを保証するための原則と実践を示している。
1. プロセスバリデーションのライフサイクル
2011年のガイドラインでは、プロセスバリデーションはライフサイクル全体にわたって実施されるべきであると強調している。具体的には、プロセスの開発段階から商業生産に至るまで、各段階でのデータ収集と評価が必要とされている。
2. 文書化された証拠
このガイドラインは、特定のプロセスがあらかじめ定めた仕様や品質特性に適合した製品を一貫して生産するための文書化された証拠を確立することを目的としている。これにより、製造プロセスの信頼性が確保される。
3. リスクベースドアプローチ
プロセスバリデーションはリスクベースドアプローチを採用し、製造プロセスの各段階での潜在的なリスクを評価し、管理することが求められる。これにより、製品の品質を維持するための効果的な戦略が構築される。
1987年と2011年のガイドラインの違い
1987年のFDAのプロセスバリデーションに関するガイドラインと2011年のガイドラインには、いくつかの重要な違いがある。以下に主な相違点を示す。
1. 定義とアプローチの変化
– 1987年のガイドラインでは、プロセスバリデーションは「特定のプロセスが、あらかじめ定めた仕様や品質特性に適合した製品を一貫して生産できることを保証するための文書化された証拠を確立すること」と定義されており、主に文書に基づくもので、手続き的な側面が強調されていた。
– 2011年のガイドラインでは、プロセスバリデーションがライフサイクル全体にわたるものであることを強調し、文書化された証拠から「科学的証拠」へと焦点が移っている。これにより、プロセスの実行中に得られるデータや情報がより重要視されるようになった。
2. バリデーションの段階
– 1987年のガイドラインでは、プロセスバリデーションは通常、少なくとも3ロットの製造を通じて行われることが求められており、プロセスの再現性を確認するための基本的な方法であった。
– 2011年のガイドラインでは、プロセスバリデーションが「三段階アプローチ」に基づいて行われることが提唱されており、プロセスの開発、商業生産、そして製品のライフサイクル全体にわたる監視が含まれ、より柔軟で科学的な方法が求められている。
3. リスク管理の強調
– 1987年のガイドラインでは、リスク管理の概念はあまり強調されていなかった。バリデーションは主にプロセスの手続き的な側面に焦点を当てていた。
– 2011年のガイドラインでは、リスクに基づくアプローチが導入され、製造プロセスの各段階でのリスク評価と管理が重要視されている。これにより、製品の品質を維持するためのより効果的な戦略が構築されるようになった。
4. 科学的根拠の必要性
– 1987年のガイドラインでは、文書化された手続きに依存していたため、実際のデータに基づく証拠は相対的に少なかった。
– 2011年のガイドラインでは、科学的根拠に基づくデータの収集と分析が重視され、プロセスの安定性や再現性を確保するための実証的なアプローチが求められている。
これらの違いは、FDAが製薬業界における品質管理の向上を図るために、より科学的でリスクに基づくアプローチを採用する必要性を認識した結果として現れている。












