設計管理における誤解
FDAは1980年代から『設計管理』について厳しい規制をかけてきた。
その理由は、医療機器はたとえ製造所で図面に従って適切に製造したとしても、そもそも設計が間違っていたら安全な医療機器にならないからである。
現状では市場における不具合の約50%が設計問題であり、そのうちの90%までもがソフトウェアの不具合である。
ところで筆者がコンサルテーションをする中で、医療機器企業の設計部門が『設計管理』について勘違や誤解または知らないということが多々ある。
『設計』と『設計管理』は異なる
まず『設計』と『設計管理』は異なると言うことである。
FDAやISO-13485が要求(規制)しているのは、『設計』ではなく『設計管理』である。
当然のことながら『設計』が出来なければ製造が出来ないわけで、各社は必ず『設計』を実施している。
どのように仕様書を作成するか、どのように図面をひくかといった『設計』行為に関しては規制要求はない。
したがって、しばしば設計書の書き方、仕様書の書き方、検証計画書の書き方などを質問されるが、言ってみればそれらは自由である。
しかしながら設計開始時には設計開発計画書を作成し、設計の各段階において設計開発計画書を遵守して『設計管理』を実施することは規制要件や国際規格で要求されている。
しかしながら現状においては、多くの医療機器企業において『設計管理』が不適切であると思われる。
設計開発計画書では設計へのインプットを定義し、『設計』のどの段階でどのようなデザインレビュを誰が実施するのかなどを詳細に記載しておかなければならない。
適切かつ詳細な設計開発計画書の作成が行われておらず、また設計開発計画書の改訂も適切に実施されていない。
また設計開発計画書の遵守も適切に実施されていない。
なお、クラスIの医療機器のほとんどは『設計管理』の適用を受けない。ただし、ソフトウェアを搭載している場合は『設計管理』の適用を受けるので注意が必要である。
『設計』の目的は『製造できるようにする』ことである
次に『設計』の目的は『製造できるようにする』ことである。
これは自明であろう。つまり製造するために『設計』するのであるから、『工程設計』も『設計』に含まれるのである。
したがって『工程設計』に関する記録類もデザインレビュにかけ、DHF(設計開発ファイル)に保存しておかなければならない。
『設計管理』において設計変更が重要であるが、いわば設計変更は製造変更である。別の言い方をすれば、製造を変更しない設計変更はあり得ないのである。
『ラベリング』も設計からのアウトプットである
『ラベル』や『ラベリング』も設計からのアウトプットであり、デザインレビュの対象であり、その記録はDHF(設計開発ファイル)に保存しておかなければならない。
ここで『ラベル』は『ラベリング』の一種である。
『ラベリング』とは、患者やユーザが目にするあらゆる印刷物を言う。
例えば、『ラベリング』に含まれるものは、取扱説明書(簡易取説等を含む)、添付文書、包装表示、サービスマニュアル、トレーニング用資料、カタログ、展示会用資料、ホームページ、製品紹介用ビデオ等である。
これらは適切に設計され、レビュされ、承認されなければならない。
ただしここで切り分けが困難なのは、どこまでがAdvertisement(宣伝)でどこからが『ラベリング』であるかということである。
品番や価格を記載しているのみではAdvertisement(宣伝)であろう。しかしながら効能・効果をうたっていれば間違いなく『ラベリング』である。
患者・ユーザの目に触れる印刷物で『ラベリング』に相当しないものは、例えば『送り状』などがある。
『梱包材』も設計からのアウトプットである
『梱包材』も設計からのアウトプットである。したがって適切に『設計管理』が実施され、デザインレビュを実施し、その記録はDHF(設計開発ファイル)に保存しておかなければならない。
精密機械等の場合、特に『梱包材』の設計は重要である。
適切にバリデーションを実施し、輸送中の衝撃、振動などから機器を守るような緩衝材・梱包材になっていなければならない。
例えば、90cmの高さから梱包した製品を落下させてみるといったバリデーションテストなどである。
FDA対応デザインコントロールの要点
☆ユーザのニーズ、意図した用途および定められた要求事項に機器が合致していることを確実にするために設計工程を管理しなければならない。
- 設計管理手順書の検証
- 合否判定基準の確立
- 検証およびバリデーション作業は、経験的ではなく、予見的であること。
- 設計バリデーション
- 機器の仕様がユーザニーズおよび意図した用途に適合していることの客観的証拠を維持・管理すること。
- 臨床評価(臨床調査、臨床試験)の実施を含み、実際あるいは模擬の使用状況での試験を含んでいること。
- 不一致事項がすべて解決されたことを証拠で示すこと。
- ソフトウェアバリデーション
- ソフトウェアによって制御している場合、CSVを実施した記録があること。
- リスク分析
- 設計バリデーション中にリスク分析を実施した記録を維持・管理すること。
- 初期生産もしくは同等品での設計バリデーションを実施した記録を維持・管理すること
- 変更管理
- 設計プロセス中になされる変更(生産前の変更)の記録を維持・管理すること。
- 最終設計の移管後の仕様変更(生産後の変更)の記録を維持・管理すること。この場合、変更要求を文書化し、インパクト(最終製品がユーザ・患者に与える影響度合い)を調査し、適切に承認がされていること。
- 全ての設計変更の検証の記録が維持・管理されていること。
- 設計の各段階またはフェーズの終わりなどで、設計レビュを実施した記録が維持・管理されていること
- 設計情報が適切に生産仕様書(DMR)に移管されたことを証明する記録を維持・管理すること
FDA査察官の視点
- 設計承認に先立ち、設計特性の容認基準が確立され文書化されているか?
- 設計インプット要求に対してデザインアウトプットのベリフィケーションがなされ、文書化されているか?
- 最終設計承認に先立ち、その設計に関するリスク分析が実施され(および文書化され)ているか?
- 設計アウトプットは機器の機能に不可欠な特性と不可欠でない特性(例:美的特性)の区別をつけているか?これらはどのように確認されているか?
- ある1つのステージの設計アウトプットが次の設計インプットになるような箇所の、設計アウトプット関連のレビュ。これらはどのように文書において明確化されているか?
- 機器の設計に対する変更が文書化され承認されていることの確認 - 実際の変更とその承認の間に日付の矛盾はないか?
- 設計変更によるリスクもしくは近接する設計インプットまたはアウトプットに対する影響はどのようにテストされているか?それはどのように文書化されているか?
- 文書化された製品設計要求、設計仕様、リスクおよび管理、テストをリンクするトレーサビリティ分析またはマトリックスはあるか?
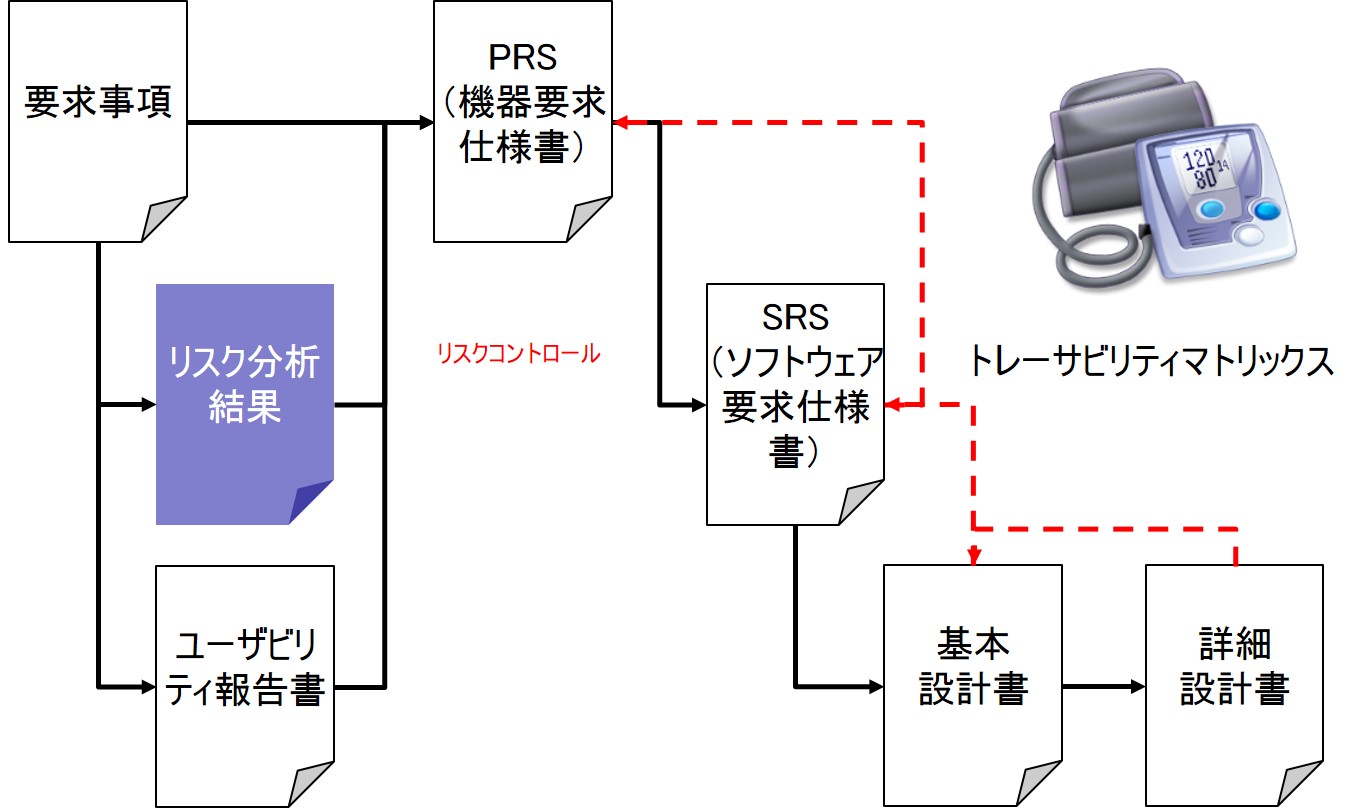
設計管理の適用開始時期とは
設計管理は、構想中または実現可能性調査中の機器には適用されない。
- 研究および実現可能性調査が終了するか、設計および開発活動と設計管理が開始されるのは何時かを記載した品質マニュアルまたは設計開発手順書を作成すること。
- 事前コンセプト研究から設計開発活動への移行は、設計開発手順書において明確に線引きしていること。
- 設計管理開始時点とは、初期設計インプット要求事項を評価・承認するデザインレビュである。
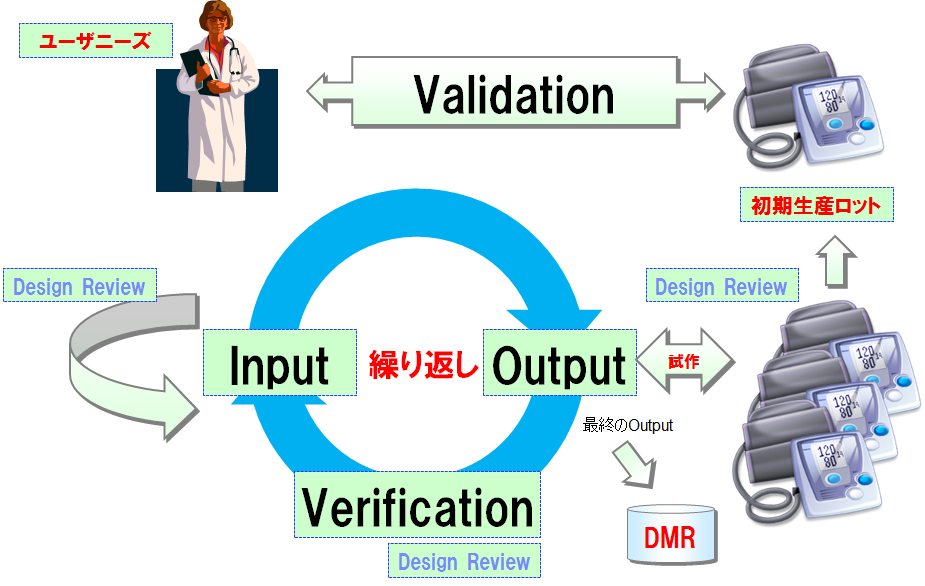
設計管理プロセス
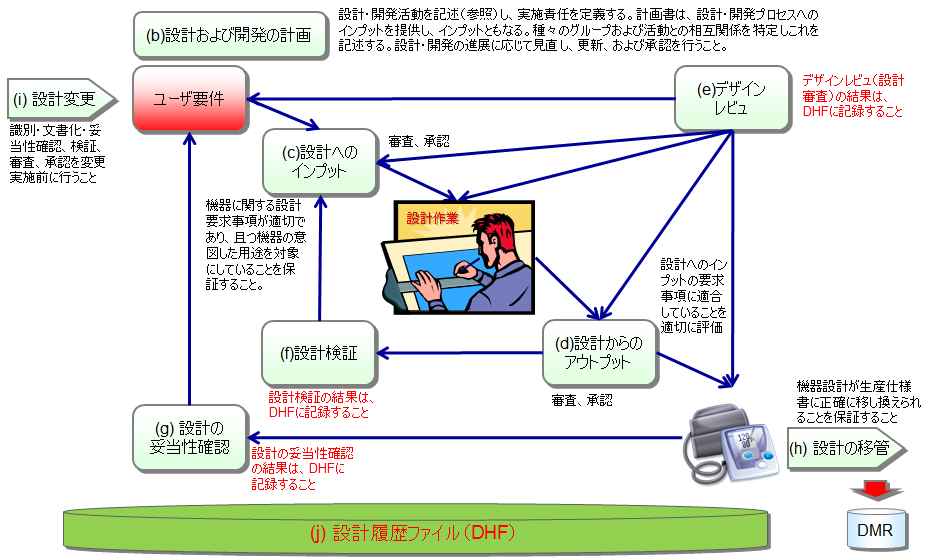
- 設計のInput(設計開発計画書、ユーザニーズ、要件仕様書、ソフトウェア仕様書等)を承認した後、設計からのOutput(図面、製品仕様書、操作説明書、製造方法、試作機等)を作成する。
- 設計作業とは、設計のInput、設計からのOutputを作成する作業に他ならない。
- デザインレビュによって、設計からのOutputをレビュする。試作機を検証する。
- 設計からのOutputが設計のInputを満たしているかどうかを、Verificationで確認する。逆も実施する。
- 設計のInputと設計からのOutputが整合しない場合、どちらかまたは双方を修正し、各々デザインレビュを実施する。再度Verificationを実施する。
- 設計のInputと設計からのOutputの整合がとれた場合、量産試作を行う。
- 量産試作機を使用して、設計Validationを実施する。
- 設計Validationに合格した最終OutputをDMRに設計移管する。
FDA査察対応設計管理手順書の作成方法
ISO-13485とFDAの要求の違い
FDAの設計管理要求(820.30)では、ISO-13485にはないDHFの管理要求がある。
FDA査察対応設計管理手順書サンプル
イーコンプライアンスでは、FDAが要求する設計管理に対応した、規程、手順書、様式のサンプルを販売しています。
ぜひご購入をご検討下さい。