
FDA 510(k)申請審査の厳格化とS/Wの信頼性保証について
最近、ME機器(Medical Engineering Device:医用電子機器)をFDAに510(k)申請したところ、ソフトウェアの信頼性について厳格な審査がなされ、FDAから問い合わせが行われるケースが増えてきた。
これまで、510(k)申請においては、FDAほほとんど審査を行ってこなかったため、批判が相次ぎ、FDAが510(k)申請審査の厳格化に関するガイダンスを発行したためである。
米国における医療機器のクラスI回収(死亡または重篤な障害の恐れがある回収)は、年間で63イベント360品目に及ぶ。つまり、毎日1台のペースで米国人が死に至るかも知れない医療機器が回収されているのである。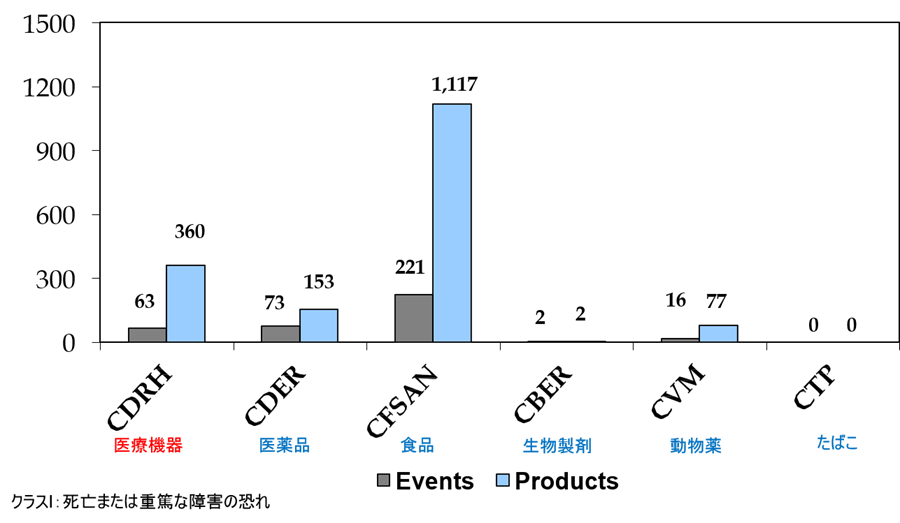
図.2014年度 米国における回収イベントと回収品目数
FDAにとって、回収数を減少させることが喫緊の課題となっている。
医療機器の回収の過半数が設計問題である。
さらにその80%以上がソフトウェアの不具合によるものである。
特に輸液ポンプのソフトウェアのバグにより、手術中に死亡事故が毎年のように発生したことからFDAの医療機器・放射線保険センター(Center for Devices and Radiological Health:CDRH)は2010年にInfusion Pump Improvement Initiativeを発行した。
FDAは、装置に含まれるすべてのソフトウェアの詳細なVerificationとValidation(V&V)を実施するよう医療機器企業に要求している。
従来は、このV&Vに使用できる唯一の手段がテストとコードレビュだった。
あいにくこれらの手法は、多数のバグを見つけるには効果があるが、ソフトウェアに起こりうる欠陥をすべて発見するという保証はできない。
テストは通常、プログラムの実行時に行われる。
プログラムの各実行はプログラムの特定のパス(条件分岐)しか通らないので、限られた数のテストでは実行パスの限られたセットしかチェックできない。
通常、これらのパスはプログラムで可能なすべてのパスのごく一部しかカバーしていない。
一方、コードレビュはレビュ担当者の技能だけが頼りで、大量のソースコードには効率的でない場合がある。
プログラムで可能なパス(またはトレース)すべてをチェックするには、プログラムのフロー解析に基づく別のアプローチが必要になる。
これを達成する方法の1つが(自動)静的解析ツールの使用である。







