
厚労省コンピュータ化システム適正管理ガイドライン(案)の考察(2010.7執筆)
ウェブセミナー
Computerized System Validationについて研究するページです。
*万が一文中に解釈の間違い等がありましても、当社では責任をとりかねます。
本文書の改訂は予告なく行われることがあります。
厚労省「コンピュータ化システム適正管理ガイドライン(案)」の考察
1. はじめに
厚生労働省は、2010年7月16日「医薬品・医薬部外品製造販売業者等におけるコン ピュータ化システム適正管理ガイドライン(案)」を発表し、パブリックコメントの募集を開始した。
パブリックコメントは、8月20日まで受け付けている。
また新ガイドライン(案)に加えて、考え方(Q&A集)も同時に掲載されている。
このガイドラインは、「コンピュータ使用医薬品等製造所適正管理ガイドライン」(平成4年2月21日薬監第11号:平成17年3月30日付薬食監麻発第0330001号により廃止)を置き換えるものである。
旧ガイドラインは、欧米の規制要件に対して、比較的内容が具体的で理解しやすく、 実用的であった。
しかしながら、IQ、OQ、PQといった検証(バリデーション)に関する記述がなく、 外国の規制当局の査察時等に苦慮するといった一面もあった。
今回は厚労省版「コンピュータ化システム適正管理ガイドライン(案)」を考察してみたい。
2. 新ガイドラインの特徴
新ガイドライン(案)を一通り読んだ印象では、以下の特徴があるようだ。
1) GMP、GQP 分野を対象としている。
2) 旧ガイドラインとGAMP 5 を折衷したような内容であ る。
3) FDA やANNEX11 等の要求事項に比べて、具体的である。
4) コンピュータ化システムの開発から、検証、運用管理及び廃棄までをライフサイクルとして定義している。
5) 多くの用語が日本語で表記されている。
6) ソフトウェアのカテゴリ分類を定義している。
7) DQ、IQ、OQ、PQ という用語を使用している。
8)「 バリデーション」という用語の定義が検証のことを意味し、狭義である。
2.1 GMP、GQP 分野を対象としている
「2. 適用の範囲」には、「このガイドラインは、GQP省令及びGMP省令が適用される業務に、コンピュータ化システムを使用する製造販売業者等に適用する。」との記載がある。
しかしながら、欧米でも同様であるように、非臨床試験や臨床試験などの研究開発においても適用されるのではないかと考えられる。
なぜならば、GLP やGCP においてもバリデーションが要求されているが、その際に拠り所とする規制要件としては、本ガイドラインが唯一であるからである。
2.2 旧ガイドラインとGAMP 5 を折衷したような内容である
新ガイドラインは、GAMP 4 やGAMP 5 を参考にしている。
規制要件に業界標準を引用することは、異例である。
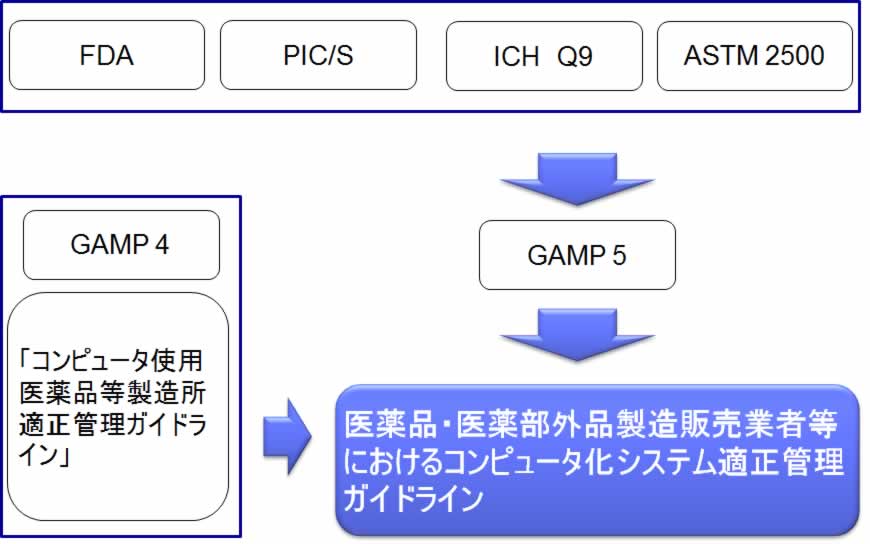
2.3 FDA やANNEX11 等の要求事項に比べて、具体的である
各成果物のコンテンツも定義している。
例えば設計仕様書の内容にも言及しているが、本来設計仕様書等は、当該供給者のQMS に従って作成されるべきである。
2.4 コンピュータ化システムの開発から、検証、運用管理及び廃棄までをライフサイクルとして定義している
旧ガイドラインでは、ライフサイクルという定義はなかった。また新ガイドラインでは、コンピュータシステムの廃棄まで範囲としていることが特徴的である。
2.5 多くの用語が日本語で表記されている
タイトルが「ガイドライン」であるにもかかわらず、条文中に「供給者」「開発責任者」「検証責任者」などの日本語表記がみられる。
供給者は、サプライヤのことであり、開発責任者は一般にプロジェクトマネージャを指すと思われる。また検証責任者はバリデーションマネージャのことであると考えられる。
一方において、「供給者アセスメント」などのカタカナ表記もみられる。
2.6 ソフトウェアのカテゴリ分類を定義している
ソフトウェアのカテゴリ分類は、GAMP に特徴的な考え方である。カテゴリ分類は、リスク評価の一つであり、数字が大きくなるほど変更の度合いが増し、不具合等のリスクが増大する。
2.7 DQ、IQ、OQ、PQ という用語を使用している
旧ガイドラインには、検証業務に関する記述がなく、したがってDQ、IQ、OQ、PQ という用語も記述がなかった。
新ガイドラインは、GAMP 4 を参考とし、これらの用語を用いている。
しかしながら、FDA やGAMP 5 では、DQ、IQ、OQ、PQという用語は使用されなくなった。
なぜならばこれらの用語は製薬業界に特徴的な呼び方であり、一般にサプライヤには理解されにくいものであるからである。
新ガイドラインは、これから発出されるにもかかわらず、このDQ、IQ、OQ、PQ という用語を使用している。この場合、すでに製薬会社がGAMP 5 等のグローバルスタンダードに対応してCSV SOPを作成していた場合、不整合が起きかねない。
いわゆるダブルスタンダードの問題が生じることが懸念される。
2.8 「バリデーション」という用語の定義が検証のことを意味し、狭義である
GAMP 4 までは、ソフトウェアの検証(すなわちテスト)のことをバリデーションと呼んだ。
新ガイドラインにおいても同じ定義である。
しかしながら、GAMP 5 は、FDA が提唱するPAT という概念およびそのインプリメントである「ASTM E2500」に整合させ、バリデーションという用語を「意図した用途への適合性の達成」と定義している。
それに呼応して、GAMP 4 のタイトルは、「GAMP Guide for Validation of Automated Systems」であったが、GAMP 5 では「A Risk-Based Approach to Compliant GxP Computerized Systems」となり、タイトルからバリデーションという用語が消えた。
ここでもまたダブルスタンダードの懸念が生じる可能性がある。
3. ガイドラインの新旧比較
新ガイドラインは、一見すると旧ガイドラインと似ているように見える。
しかしながら、その内容は大きく異なる。(表1 参照)
| コンピュータ使用医薬品等製造所適正管理ガイドライン | 医薬品・医薬部外品製造販売業者等におけるコンピュータ化システム適正管理ガイドライン(案) |
|---|---|
| 第1 目的第2 適用の範囲 | 1. 総則 1.1 目的 1.2 コンピュータ化システムの取扱い 組織・役割に応じた責任と権限の明確化 厚労省ER/ES指針を遵守すること 回顧的なバリデーションの実施 1.3 カテゴリ分類 2. 適用の範囲 3. コンピュータ化システムの開発、検証及び運用の手順等に関する文書の作成 |
| 第3 開発業務1. 開発検討段階(1) 開発段階の責任体制の確立(2) 開発マニュアルの作成(3) 開発計画書の作成2. システム設計段階(1) システム設計書の作成(2) システム設計書の確認3. プログラム設計段階(1) プログラム仕様書の作成(2) プログラム仕様書及びプログラムの確認(3) プログラムテストの実施4. システムテスト段階(1) システムテスト実施計画書の作成(2) システムテストの実施5. 設置・運用テスト段階(1) 設置計画書の作成(2) ハードウェアの設置(3) 運用テスト実施計画書の作成(4) 運用テストの実施 | 4. 開発業務4.1 開発計画書の作成4.2 要求仕様書の作成4.3 システムアセスメント4.4 機能仕様書の作成4.5 設計仕様書の作成4.5.1 ハードウェア設計仕様4.5.2 ソフトウェア設計仕様4.6 プログラムの作成及びプログラムテスト4.6.1 プログラムの作成4.6.2 プログラムテストの計画及び実施4.7 システムテスト4.7.1 システムテスト計画書の作成4.7.2 システムテストの実施4.8 受入試験 |
| 5. 検証業務5.2 バリデーション全体計画書の作成5.2.1 設計時適格性評価計画書の作成5.2.2 設計時適格性評価の実施5.2.3 設計時適格性評価報告書の作成5.3 据付時適格性(IQ)5.4 運転時適格性評価(OQ)5.5 性能適格性評価(PQ)5.6 適格性評価の一部省略と引用5.7 バリデーション総括報告書の作成 | |
| 第4 運用管理業務1. 総則(1) 運用管理段階の責任体制の確立(2) 運用管理手順書の作成(3) システムの変更2. ハードウェアの操作3. 保守点検事項の実施4. 事故発生時の対応5. セキュリティ管理の実施6. 自己点検の実施 | 6. 運用管理業務6.1 運用管理における責任体制の確立6.2 運用管理の手順に関する文書の作成6.3 コンピュータシステムの操作6.4 保守点検事項の実施6.5 セキュリティ管理の実施6.6 バックアップ及びリストア6.7 変更の管理6.8 逸脱の管理6.9 教育訓練6.9.1 教育訓練計画の作成6.9.2 教育訓練の実施 |
| 7.自己点検7.1 自己点検の実施7.2 改善措置の実施 | |
| 8. コンピュータシステムの廃棄8.1 コンピュータシステムの廃棄計画書の作成8.2 コンピュータシステムの廃棄記録の作成 | |
| 第5 文書及び記録の保存管理 | 9. 文書及び記録の管理 |
| 10. 用語集 |
表1 ガイドライン新旧比較
以下に、新ガイドラインで追加となった、主要な要件を記載する。
1)「 コンピュータ化システム管理規定」等の作成
2) 組織・役割に応じた責任と権限の明確化
3) 厚労省ER/ES 指針の要件の遵守
4) 回顧的なバリデーションの実施
5) システム台帳の作成
6) 要求仕様書の作成
7) システムアセスメントの実施
・ソフトウェアのカテゴリ分類
・製品品質に対するリスクアセスメント
・供給者アセスメント
8) 検証業務(DQ、IQ、OQ、PQ)の実施
9) 改善措置の実施
10) コンピュータシステムの廃棄
11) 業務の継続性のための要件、障害対策の要件、データのバックアップ、アクセス制限、アクセス記録等
3.1 「コンピュータ化システム管理規定」等の作成
新ガイドラインでは、「コンピュータ化システム管理規定」等のCSV SOP を作成することを求めている。
一般に、CSV SOP は、最上位にCSV Policy を作成し、経営者が承認する。
Policy には、コンピュータ化システムの開発、検証、運用及び廃棄に関する基本方針や、体制および役割と責任を記載
しなければならない。
またPolicy の下位には、手順書と各種チェックリストを作成しなければならない。
手順書には、作成すべき文書及びその管理方法や業務完了の確認及び承認の手続きを記載する。
チェックリストは、システムアセスメントや供給者監査の際に必要となるので準備が必要である。
3.2 組織・役割に応じた責任と権限の明確化
CSV を実施するにあたっての組織および役割と責任を明確にしておかなければならない。
ただし、品質保証に関する責任体制は、GQP 省令とGMP省令で異なるため、双方にまたがって体制を定義する場合に
は、注意が必要である。
3.3 厚労省ER/ES 指針の要件の遵守
「医薬品等の承認又は許可等に係る申請等に関する電磁的記録・電子署名利用のための指針」(いわゆる厚労省ER/ES指針)は、平成17年4月1日に局長通知として発出された。
発出からすでに5 年以上が経過している。
ANNEX 11(改定版ドラフト) には、電子記録・電子署名に関する要件があるため、整合性をとるために本要件が入れられたものと推察する。
米国でも1997 年に21 CFR Part 11 が発効され、これまで多くの製薬会社がその対応に苦慮してきたことは周知の事
実である。
日本版のPart11 とも呼ばれる、厚労省ER/ES 指針に準拠するためには、相応の努力が必要となると思われる。
3.4 回顧的なバリデーションの実施
PIC/S のガイダンスには、回顧的バリデーションの記載がみられるため、整合性をとったものと推察する。
回顧的バリデーションは、旧ガイドライン等に従ってバリデーションしていないシステムについて、回顧的に品質補償
を行うといったものである。
現在稼働中にシステムや設備に関して、各々品質保証ができているかを再度精査しておかなければならない。
この機会にシステムの棚卸を行っておくべきである。
ここにおいて「回顧的バリデーション」ではなく、「回顧的なバリデーション」であることに注意が必要である。
新ガイドライン(案)とともに発表された「考え方」の問31 には、「回顧的なバリデーションを実施する場合、具体的
にはどのように実施すればよいのか」という質問があり、以下のように回答している。
「回顧的なバリデーションについては、当該システムの開発時の仕様書などの文書類や記録類に遡って、その適格性を
検証する方法や、現在の使用目的に適合した要求仕様やそれに準じる文書との適格性を確認する方法等が考えられるが、
適格性の確認にあたっては、現在の運用における記録類の照査や定期的レビューの結果を利用してもよい。」
つまり、回顧的なバリデーションには3 つの方法が考えられる。
1) 過去の仕様書を精査する。場合によってはそれらを再作成する。
2) 現在のシステムの利用方法に適合するよう、ユーザ要求仕様書を新規作成する。
3) 稼働後の実データを照査し、問題がないことを確認する。
上記のうち、3) が本来の回顧的バリデーションである。
新規バリデーション(Prospective Validation)の場合、システムはまだ稼働していないため、実データはなく、テスト
データを用いた仮説検証型のテストを実施することになる。
これに対して、回顧的バリデーション(Retorospective Validation)では、すでに稼働していることから、テストデー
タではなく、より現実的な実データを検証することにより、当該システムの信頼性を確認することができる。したがって
一般に回顧的バリデーションでは、テストを実施しない。
しばしば、稼働しているシステムを再度テストすることを回顧的バリデーションと呼んでいるのを耳にするが、正しく
は再バリデーション(Re-Validation)と呼ぶ。
3.5 システム台帳の作成
システム台帳は、一般に英語ではシステムインベントリと呼ばれる。
これは施設内や工場内に設置されているコンピュータ化システムを一覧表にまとめたものである。
欧米の当局は、以前からこのシステムインベントリの作成を義務付けてきた。
コンピュータ化システムの査察を受ける際には、このシステムインベントリを提示し、当該施設内や工場内にどのよう
なコンピュータ化システムが存在するのかを提示できなければならないからである。
システムインベントリには、H/W の名称、S/W の名称とバージョン、作成したSOP やバリデーションの記録を記載
しておかなければならない。
またシステムリスクアセスメントによって判定された、リスクについても記載しておく必要がある。
初回にシステムインベントリを作成する際は、固定資産台帳などを参考にすると良いだろう。
ここで注意が必要なことは、個人にパソコンにインストールされているExcel 等のアプリケーションである。
Excel は一般の量販店でも購入できる安価なアプリケーションであるため、見落としがちであるが、万が一、品質試
験の記録、出荷判定の記録、製造記録等のリスクの高い記録類等を保持していたり、管理している場合は要注意である。
システムの価格にかかわらず、扱う電子記録の重要性やリスクに応じた対応が重要といえる。
システムインベントリを作成する際には、当該部門におけるExcel の利用についても徹底的に調査しておくことが肝心
である。
3.6 要求仕様書の作成
要求仕様書は、一般的にはユーザ要求仕様書と呼ばれる。
旧ガイドラインでは、ユーザ要求仕様書の作成が要求されていなかった。
しかしながら、欧米の規制要件やGAMP 等では、ユーザ要求仕様書の作成は必須である。
なぜならば、バリデーションとは、ユーザの要求を満足するようコンピュータ化システムを開発、導入、運用すること
であるからである。
品質保証の基本原則は、あらかじめ定めた仕様や品質が存在し、最終的にそれらを満たしたことを証明する証拠が存在
しなければならないのである。
3.7 システムアセスメントの実施
システムアセスメントでは、以下の3 つを実施するように求めている。
1) ソフトウェアのカテゴリ分類
あらかじめソフトウェアのカテゴリ分類を実施しなければならない。
ソフトウェアのカテゴリ分類は、GAMP でよく知られている。
欧米の規制要件には、このソフトウェアのカテゴリ分類はなく、本ガイドラインに特徴的である。
しかしながら、ソフトウェアのカテゴリ分類は、サプライヤと協議し、設計が固まらないと決定できないという問題があ
り、開発業務の初期段階では決定は困難である。
ソフトウェアのカテゴリ分類の詳細については、GAMP 5 を学習しておく必要がある。
2) 製品品質に対するリスクアセスメント
当該コンピュータ化システムが生産する薬剤等の製品のリスクを判定しなければならない。
すなわち、当該製品の品質に異常があった際の患者に与える健康被害のリスクをあらかじめ調査しておくのである。
リスクアセスメントに関する詳細は、ICH Q9 を学習しておかなければならない。
3) 供給者アセスメント
コンピュータ化システムの品質は、当該サプライヤによるところが大きい。
したがって、サプライヤの調査および選定が重要であるといえる。
サプライヤの選定にあたっては、サプライヤオーディットを実施しなければならない。
サプライヤオーディットは、Ad-hoc に実施すべきではなく、チェックリストを用いて、十分に計画した上で実施する必要がある。
3.8 検証業務(DQ、IQ、OQ、PQ)の実施
旧ガイドラインでは、検証業務に関する記述がなかった。
新ガイドラインでは、GAMP 4 に従い、また現在のところ欧米を中心に多く実施されているDQ、IQ、OQ、PQ と呼ば
れる検証方法を用いている。
しかしながら、前述したとおり、最新の欧米の規制要件やGAMP 5 では、DQ、IQ、OQ、PQ という用語を使用しなくなったため、今後日本企業においては、ダブルスタンダードの問題が懸念される。
3.9 改善措置の実施
改善措置についても、新ガイドラインに新しく追加された要求事項である。
FDA は、2004 年から、CAPA(Corrective Actions; Preventive Actions:予防措置・是正措置)に関する査察を
強化した経緯がある。
3.10 コンピュータシステムの廃棄
コンピュータシステムの廃棄についても、新ガイドラインで追加となった。
コンピュータシステムを廃棄する際には、H/W とS/W は廃棄しても構わないが、当該システムの操作手順書、運用マ
ニュアル等の手順書類や、CSV の記録等は捨て去ってはならない。
また当該コンピュータシステムが保持している、電子記録や電子署名は、正確かつ完全に安全な場所にアーカイヴして
おかなければならない。
ここで完全とは、監査証跡等のメタデータも含めて、移行しておくという意味である。
そのうえで、電子記録・電子署名は、査察が実施された際に、査察官に提示できるよう、検索と印刷の機能を保有して
いなければならない。
3.11 業務の継続性のための要件、障害対策の要件、データのバックアップ、アクセス制限、アクセス記録等
新ガイドライン(案)とともに発表された「考え方」のQ33 には、「コンピュータシステムが備える要件」について
の記載がある。
「本ガイドラインの対象となるコンピュータシステムには、どのような要件を備える必要があるのか。」という問い
に対し、以下のような要件を回答している。
• ER/ES 指針の要件
• 真正性、見読性、保存性確保のための要件
• 業務の継続性のための要件
• セキュリティ確保のための要件
• バックアップ、アクセス制限、アクセス記録等に関する要件
• バックアップメディアに求められる要件
• メディアの保存方法の要件
これらの要件もANNEX 11 との整合性を持たせたものであると推察されるが、ではなぜガイドライン本体に記載して
いないのであろうか。
「考え方」のQ34 では、業務継続性とはについて以下のように回答している。
業務継続性とは、故障やシステムトラブルによる、業務の中断を避けるため、回避のための措置を講じたり、トラブル
が発生しても、当該業務が継続可能な措置を用意するなどの対応を行うことである。
業務の継続性確保のための要件とは
• 天災や人為的妨害を考慮した設置条件の設定
• データのバックアップ(バックアップの方法や保存方法等)
• 同一の代替機をあらかじめ用意
• 予めマニュアルによる代替手順を規定しておく
• 迅速なシステムの復旧のための措置の手順
• 定期的にデータのバックアップを保存
などである。
業務継続性の必悪性は、リスクアセスメントの結果等を考慮して当該リスクに応じて決定することになる。
4. 新ガイドライン対応の留意点
新ガイドラインに対応するために留意しなければならないと思われる事項を記載する。
1.ダブルスタンダードへの対応
上述した通り、グローバルのCSVに関する用語と新ガイドラインの用語では、その使い方や定義に違いがある。したがってグローバル企業にとっては、ダブルスタンダードに対応しなければならないことになる。
2.本ガイドラインに代わる方法を用いても可としているが、日本の査察においては、本ガイドラインをもとに実施されるので、注意が必要である。
3.ソフトウェアのカテゴリ分類については、GAMP 5を参考にしているため、GAMP 5をよく理解しておかなければならない。またソフトウェアのカテゴリ分類の方法は、各社で決定し、コンピュータ化システム管理規定等に記載しておかなければならない。
4.定義に記載されていない用語
GAMP、ソフトウェアカテゴリ、リスク、システム台帳等の用語は定義されていない。
5.リスクアセスメントについては、ICH-Q9をよく理解して対応する必要がある。
5. 新ガイドライン条文解釈
1.総則
| 1.総則 1.1 目的このガイドラインは、「コンピュータ使用医薬品等製造所適正管理ガイドライン」(平成4年2月21日薬監第11号:平成17年3月30日付薬食監麻発第0330001号により廃止)に代わるものとして、「医薬品及び医薬部外品の製造管理及び品質管理の基準に関する省令」(平成16年厚生労働省令第179号。以下「GMP省令」という。)の適用を受ける医薬品又は医薬部外品を製造販売する製造販売業者又は製造する製造業者等(以下「製造販売業者等」という。)が、「医薬品、医薬部外品、化粧品及び医療機器の品質管理の基準に関する省令」(平成16年厚生労働省令第136号。以下「GQP省令」という。)及びGMP省令に基づく業務を行うためのコンピュータ化システムについて、これを開発する際に必要な事項、これを検証するバリデーションに関する事項及び運用管理に関する遵守事項(バリデートされた状態の維持や廃棄に関する事項等)を定め、GQP省令及びGMP省令の適正な実施の確保を図ることを目的とする。 |
本ガイドラインは、旧ガイドラインである「コンピュータ使用医薬品等製造所適正管理ガイドライン」を置き換えるものである。
| 1.総則 1.1 目的(続き)このガイドラインにおいては、コンピュータ化システムの開発から、検証、運用管理及び廃棄までの流れを総合してコンピュータ化システムのライフサイクルという。ライフサイクル全体の構成を別紙1「コンピュータ化システムのライフサイクルモデル」に示す。また、このガイドラインに示した管理方法は標準的な例を示したものであり、これに代わる方法で、それが同等又はそれ以上の目的を達成できるものである場合には、その方法を用いても差し支えない。 |
「代わる方法」とは、GAMP 5等を指す。
しかしながら、日本の査察においては、本ガイドラインをもとに実施されるので、注意が必要である。
| 1.2 コンピュータ化システムの取扱いこのガイドラインは、GQP省令及びGMP省令に関連するシステム並びに相互に連携したコンピュータ化システムを対象として取り扱うこととしているため、GQP省令やGMP省令における組織・役割に応じた表現を用いていないが、バリデーションや変更・逸脱の管理など、GMP省令においては品質部門等の承認が必要であり、GQP省令においては品質保証部門による管理体制の中で進めなければならない。従って、製造販売業者等において組織の形態や該当するシステムの範囲を考慮して各々の組織・役割に応じた責任と権限を3.に規定する「コンピュータ化システムの開発、検証及び運用の手順等に関する文書」の中に明確にすることが必要である。 |
組織・役割に応じた責任と権限を明確にする必要がある。
| 1.2 コンピュータ化システムの取扱い(続き)また、このガイドラインの対象となるコンピュータ化システムは「医薬品等の承認又は許可等に係る申請等に関する電磁的記録・電子署名利用のための指針」(平成17年4月1日薬食発第0401022号)及び「薬事法及び採血及び供血あつせん業取締法の一部を改正する法律の施行に伴う医薬品,医療機器等の製造管理及び品質管理(GMP/QMS)に係る省令及び告示の制定及び改廃について」 (平成17年3月30日薬食監麻発第0330001号)第3章第3 35.「その他(電磁的記録等について)」の要件を備える必要がある。 |
厚労省ER/ES指針に準拠すること。
| 1.2 コンピュータ化システムの取扱い(続き)なお、このガイドラインの施行日以前に開発又は運用が開始されているシステムであって、「コンピュータ使用医薬品等製造所適正管理ガイドライン」に示された方法又はそれに代わる適切な方法で開発、検証及び運用等が行われていないシステムについては、回顧的なバリデーション等により、当該システムの適格性を確認する必要がある。 |
回顧的なバリデーション:旧ガイドラインに従って開発・管理されているのが前提だが、転用されたシステム等の対応が必要。
| 1.3 カテゴリ分類このガイドラインの適用を受けるコンピュータ化システムについては、開発、検証及び運用の各段階において実施する内容を決定(「4.3システムアセスメント」を参照)するために、システムを構成するソフトウェアの種類に応じて、あらかじめソフトウェアカテゴリを決定するものとする。カテゴリ分類の基準及びカテゴリ毎の一般的対応の例を別紙2「カテゴリ分類表」に示した。 |
本ガイドラインでは、GAMPと同様、ソフトウェアをカテゴリ分けしている。
カテゴリはすなわちソフトウェアのリスクの分類である。カテゴリの数字が大きくなるほど、ユーザによる変更の度合いが大きく、ソフトウェアに欠陥が含まれる可能性が増大する。
リスクアセスメントの結果としてより簡便な取扱を行なっても差し支えないとする合理的な根拠を示すことができるようにしなければならない。
2.適用の範囲
| 2. 適用の範囲このガイドラインは、GQP省令及びGMP省令が適用される業務に、コンピュータ化システムを使用する製造販売業者等に適用する。このガイドラインの対象となるコンピュータ化システムの例として、以下が考えられる。(1) 医薬品、医薬部外品の市場への出荷の可否の決定に係るシステム及び市場への出荷に係る記録を作成、保存管理するためのシステム(2) 製造指図書、製造に関する記録等を作成及び保存管理するためのシステム(3) 製造工程を制御又は管理するためのシステム及びその管理データを保存管理するためのシステム(4) 原材料及び製品(製造の中間工程で造られるものを含む。以下同じ。)の保管、出納等の生産を管理するシステム(5) 品質試験のための機器を制御・管理するシステム並びに品質試験結果及び管理データを保存管理するためのシステム(6) 空調、製造用水製造設備など、製品の品質に重大な影響を及ぼす可能性のある製造支援設備・施設を制御又は管理するためのシステム及びその管理データを保存管理するためのシステム(7) 文書(手順書類、品質標準書、製品標準書等)を作成、承認、保存管理するためのシステム |
3.コンピュータ化システムの開発、検証及び運用の手順等に関する文書の作成
| 3. コンピュータ化システムの開発、検証及び運用の手順等に関する文書の作成製造販売業者等はコンピュータ化システムの開発、検証及び運用にあたっては、あらかじめ、その手順等に関する文書(以下「コンピュータ化システム管理規定」という。)を定めるものとする。コンピュータ化システム管理規定は、原則として次の事項を記載するものとする。(1) コンピュータ化システムの開発、検証及び運用に関する基本方針-目的-適用範囲-システム台帳の作成-基本的な考え方・ソフトウェアのカテゴリ分類・製品品質に対するリスクアセスメント・供給者アセスメント・開発、検証及び運用段階で実施すべき項目等(2) 開発業務、検証業務及び運用管理業務における責任体制と役割(3) 開発業務、検証業務及び運用管理業務で作成すべき文書及びその管理方法(4) 開発業務、検証業務及び運用管理業務の業務完了の確認及び承認の手続き |
コンピュータ化システム管理規定は企業全体で統一した文書として制定する場合もあるし、個々の部門で個別に制定する場合もある。
統一した文書を制定する場合であっても、GQPやGMPの基準に沿って文書の承認を行うことが必要である。
個々に制定する場合は、必要に応じて部門間での基本的取扱いの整合性の確保と、部門間での役割分担と連携の方法について記載しておくことが必要である。
4.開発業務
| 4. 開発業務 4.1 開発計画に関する文書の作成製造販売業者等は、開発計画に関する事項を記載した文書(以下「開発計画書」という。)を作成する。開発計画書には、原則として次の事項を記載するものとする。(1) 開発の目的(2) 開発条件(3) 開発体制-組織-責任者・開発責任者・検証責任者(4) 開発スケジュール |
| 4.2 要求仕様に関する文書の作成開発責任者はコンピュータ化システムに求められている事項を記載した文書(以下「要求仕様書」という。)を作成する。要求仕様書には原則として次の事項を記載するものとする。(1) 適用する法規制等(2) ハードウェアの概要(3) 要求機能-システムの機能の概要-運用要件の概要-性能要件の概要-障害対策機能の概要-機密保護機能の概要(セキュリティ)(4) データ-入出力情報の項目の一覧-保存方法(5) インターフェース(関連設備及び他システム等)(6) 環境-設置条件-システムの配置(7) 電源、接地等の据付条件 |
| 4.3 システムアセスメント開発責任者は開発、検証及び運用の各段階にて実施すべき項目等を決定し、それぞれの内容を定めるために、コンピュータ化システム管理規定に基づき以下の事項を実施する。(1) ソフトウェアカテゴリ分類(2) 製品品質に対するリスクアセスメント(3) 供給者アセスメント |
本ガイドラインでは、GAMPと同様、ソフトウェアをカテゴリ分けしている。
カテゴリはすなわちソフトウェアのリスクの分類である。カテゴリの数字が大きくなるほど、ユーザによる変更の度合いが大きく、ソフトウェアに欠陥が含まれる可能性が増大する。
リスクアセスメントの結果としてより簡便な取扱を行なっても差し支えないとする合理的な根拠を示すことができるようにしなければならない。
| 4.4 機能仕様に関する文書の作成開発責任者は、供給者に要求仕様書に記載された要件に対応した具体的なコンピュータ化システムの機能と性能を記述した機能仕様を記載した文書(以下「機能仕様書」という。)を作成させ、承認するものとする。 |
| 4.5 設計仕様に関する文書の作成開発責任者は、供給者に機能仕様に基づいてコンピュータ化システムの詳細機能を記述した設計仕様に関する文書(以下「設計仕様書」という。)を作成させ、承認するものとする。設計仕様書には、原則として次の事項を記載するものとする。4.5.1 ハードウェア設計仕様(1) ハードウェア構成(2) ハードウェアリスト及び仕様(3) インターフェース(4) 入出力信号の詳細(5) 環境-設置の詳細条件-システム機器の配置(6) 電源、接地等の据付条件4.5.2 ソフトウェア設計仕様(1) 入出力情報の詳細(2) ファイル及びデータ構造(3) データ処理の詳細(4) 機能・モジュールの構成(5) インターフェースの詳細(6) 選択したパッケージソフトウェア |
| 4.6 プログラムの作成及びプログラムテスト開発責任者は、必要に応じて、供給者にプログラム作成及びプログラムテストを実施させるものとする。プログラム作成及びプログラムテストには、以下の内容が含まれるものとする。4.6.1 プログラムの作成(1) 供給者は、プログラムの仕様に関する文書(以下「プログラム仕様書」という。)を設計仕様書に従って作成するものとする。(2) 供給者はプログラムをプログラム仕様書どおりに作成するものとする。4.6.2 プログラムテストの計画及び実施(1) 供給者は、プログラムテスト方法、プログラムテスト結果の判定方法及び判定基準を記載したプログラムテストの計画に関する文書(以下「プログラムテスト計画書」という。)を作成するものとする。(2) 供給者は、プログラムテスト計画書に基づき、プログラムテストを実施し、その結果を記録するものとする。(3) 供給者は、プログラムテストの結果の適否を判定するものとする。 |
| 4.7 システムテスト開発責任者は、必要に応じて供給者にシステムテストを実施させるものとする。システムテストには以下の内容が含まれるものとする。4.7.1 システムテストの計画に関する文書の作成供給者はシステムテストにあたっては、システムテストの計画に関する文書(以下「システムテスト計画書」という。)を作成するものとする。システムテスト計画書には、原則として次の事項を記載するものとする。(1) システムテストの実施環境(テスト時のハードウェアの設置状況及びソフトウェア構成等をいう。)(2) システムテストの項目及び使用するテストデータ(3) システムテストの方法及び結果の確認方法(4) システムテストの判定基準(5) システムテストのスケジュール(6) システムテストを実施する場合の実施体制4.7.2 システムテストの実施(1) 供給者は、システムテスト計画書に基づいてシステムテストを実施し、その結果(システムテストの実施時に発生したトラブルの内容及びその措置内容を含む。)を記録するものとする。(2) 供給者は、システムテストの結果の適否を判定する。この場合において、システムテストの結果の適否の判定事項は、原則として次のとおりとする。-機能(機能仕様書及び設計仕様書に規定されたとおりに機能するか等)-性能(機能仕様書及び設計仕様書で期待された応答性等を確保しているか等) |
| 4.8 受入試験開発責任者は、システムの機能及び性能の全てあるいは一部が要求仕様を満足していることを確認するために供給者に受入試験を実施させる。受入試験には、供給者の工場出荷前に機能及び性能を確認するテスト(工場出荷試験、以下「FAT」という。)並びにこれらシステム設置場所等における受け入れ時に機能及び性能を確認するテスト(現地受入試験、以下「SAT」という。)があり、適宜選択し実施させる。受入試験の結果は開発責任者が承認する。 |
5.検証業務
| 5. 検証業務 5.1 バリデーションの全体計画に関する文書の作成検証担当者は、コンピュータ化システム管理規定に基づき、システムの検証を行う場合には、実施するバリデーションの全体計画に関する文書(以下「バリデーション計画書」という。)を作成するものとする。なお、バリデーション計画書は「4.3システムアセスメント」により実施した評価結果等に基づき作成する。バリデーション計画書は検証責任者の承認を得るものとする。なお、検証業務は開発業務と併行して行われることもあるため、バリデーション計画書は開発段階の適切な時期に作成する。また、「6.7変更の管理」においてバリデーションが必要となった場合は、変更の状況にあわせて適宜バリデーション計画書を作成すること。バリデーション計画書には、原則として次の事項を記載するものとする。また、必要な場合には詳細なリスクアセスメント、供給者監査等の計画についても記載すること。 |
| 5. 検証業務 5.1 バリデーションの全体計画に関する文書の作成(続き)(1) 目的(2) システム概要(3) 責任体制と役割 -組織 -検証責任者(4) 適用する法規制等(5) バリデーション方針-バリデーションの範囲及びバリデーションとして実施すべき項目等(6) スケジュール(7) バリデーション実施時の変更・逸脱の管理に関する手順 |
| 5.2 設計時適格性評価(DQ) 5.2.1 設計時適格性評価の計画に関する文書の作成検証担当者は、設計時適格性評価の計画に関する文書(以下「設計時適格性評価計画書」という。)を作成し、検証責任者の承認を得るものとする。設計時適格性評価計画書には、原則として次の事項を記載するものとする。(1) 設計時適格性評価に係る文書名(2) 具体的な確認の方法(3) 設計時適格性評価における判定基準(4) スケジュール(5) 責任者及び担当者の氏名5.2.2 設計時適格性評価の実施(1) 検証担当者は、設計時適格性評価計画書に基づいて評価を実施し、その結果を記録するものとする。(2) 検証責任者は、設計時適格性評価の結果の適否を判定するものとする。5.2.3 設計時適格性評価の報告に関する文書の作成検証担当者は、設計時適格性評価の報告に関する文書(以下「設計時適格性評価報告書」という。)を作成し、検証責任者の承認を得るものとする。設計時適格性評価報告書には、原則として次の事項を記載するものとする。(1) 設計時適格性評価に係る文書名(2) 評価結果と是正措置(3) 責任者及び担当者の氏名 |
| 5.3 据付時適格性評価(IQ) 5.3.1 据付時適格性評価の計画に関する文書の作成検証担当者は、ハードウェア及びソフトウェアの据付時適格性評価の計画に関する文書(以下「据付時適格性評価計画書」という。)を作成し、検証責任者の承認を得るものとする。据付時適格性評価計画書には原則として次の事項を記載するものとする。(1) 据付時適格性評価に係る文書名(2) ハードウェア構成及び設置場所(3) ハードウェアの提供業者が推奨する温度、湿度、振動等の環境条件(4) 電源、接地等の設置条件(5) 通信、入出力に関する仕様(6) ハードウェアの設置の適否の確認方法(7) ソフトウェアのインストールの適否の確認方法(8) 据付時適格性評価における判定基準(9) スケジュール(10) 責任者及び担当者の氏名5.3.2 据付時適格性評価の実施(1) ハードウェアの設置-検証担当者は据付時適格性評価計画書に基づいて、ハードウェアが適切に設置されていることを確認し、その結果を記録するものとする。-検証責任者は、ハードウェアの設置の適否を判定するものとする。(2) ソフトウェアのインストール-検証担当者は基本ソフトウェアを含め、適切にインストールされていることを確認し、その結果を記録するものとする。-検証責任者は、ソフトウェアのインストールの結果の適否を判定するものとする。5.3.3 据付時適格性評価の報告に関する文書の作成検証担当者は、据付時適格性評価の報告に関する文書(以下「据付時適格性評価報告書」という。)を作成し、検証責任者の承認を得るものとする。据付時適格性評価報告書には、原則として次の事項を記載するものとする。(1) 据付時適格性評価に係る文書名(2) 評価結果と是正措置(3) 責任者及び担当者の氏名 |
| 5.4 運転時適格性評価(OQ) 5.4.1 運転時適格性評価の計画に関する文書の作成検証担当者は、運転時適格性評価の計画に関する文書(以下「運転時適格性評価計画書」という。)を作成し、検証責任者の承認を得るものとする。運転時適格性評価計画書には原則として次の事項を記載するものとする。(1) 運転時適格性評価に係る文書名(2) システムの稼働環境における機能の確認方法(3) 運転時適格性評価における判定基準(4) スケジュール(5) 責任者及び担当者の氏名5.4.2 運転時適格性評価の実施(1) 検証担当者は、運転時適格性評価計画書に基づいて評価を実施し、その結果を記録するものとする。(2) 検証責任者は、運転時適格性評価の結果の適否を判定するものとする。5.4.3 運転時適格性評価の報告に関する文書の作成検証担当者は、運転時適格性評価の報告に関する文書(以下「運転時適格性評価報告書」という。)を作成し、検証責任者の承認を得るものとする。運転時適格性評価報告書には、原則として次の事項を記載するものとする。(1) 運転時適格性評価に係る文書名(2) 評価結果と是正措置(3) 責任者及び担当者の氏名 |
| 5.5 性能適格性評価(PQ) 5.5.1 性能適格性評価の計画に関する文書の作成検証担当者は、性能適格性評価の計画に関する文書(以下「性能適格性評価計画書」という。)を作成し、検証責任者の承認を得るものとする。性能適格性評価計画書には原則として次の事項を記載するものとする。(1) 性能適格性評価に係る文書名(2) システムの稼働時における機能及び性能の確認方法(3) 性能適格性評価における判定基準(4) スケジュール(5) 責任者及び担当者の氏名5.5.2 性能適格性評価の実施(1) 検証担当者は、性能適格性評価実施計画書に基づいて、性能適格性評価を実施し、その結果を記録するものとする。(2) 検証責任者は、性能適格性評価の結果の適否を判定するものとする。5.5.3 性能適格性評価の報告に関する文書の作成検証担当者は、性能適格性評価の報告に関する文書(以下「性能適格性評価報告書」という。)を作成し、検証責任者の承認を得るものとする。性能適格性評価報告書には、原則として次の事項を記載するものとする。(1) 性能適格性評価に係る文書名(2) 評価結果と是正措置(3) 責任者及び担当者の氏名 |
| 5.6 適格性評価の一部省略と引用(1) 「5.4 運転時適格性評価(OQ)」における検証内容、環境、条件などが「5.5 性能適格性評価(PQ)」の内容と差がない場合は運転時適格性評価を省略しても差し支えないものとする。但しその場合、省略の旨を「バリデーション計画書」若しくは「性能適格性評価計画書」又はいずれかの報告書に明記すること。(2) FAT又はSATを行った場合等、その確認の方法及び記録が検証責任者によって適切と認められる場合には、適格性評価にあたって、その結果を引用しても差し支えないものとする。 |
| 5.7 バリデーションの全体報告に関する文書の作成検証担当者は、バリデーションの各段階の結果及び総合評価をまとめたバリデーションの全体報告に関する文書を作成し、検証責任者の承認を得るものとする。 |
6.運用管理業務
| 6. 運用管理業務 6.1 運用管理における責任体制製造販売業者等はコンピュータ化システムの運用管理に関する役割と責任を定めるものとする。 |
| 6.2 運用管理の手順に関する文書の作成製造販売業者等はコンピュータ化システムの運用管理の手順に関する文書(以下「運用管理手順書」という。)を作成するものとする。運用管理手順書には原則として次の事項を記載するものとする。但し、GQP省令又はGMP省令に関する手順書に基づき管理を行う項目については、その旨を記載すること。(1) 運用に関する責任体制と役割-組織-運用責任者(2) コンピュータ化システムの操作に関する事項コンピュータ化システムの操作の手順に関する文書(標準操作手順書)をコンピュータ化システムごとに作成し、それに基づき操作するものとする。(3) 保守点検管理に関する事項① 日常点検事項② 定期点検事項③ 保守点検を専門業者に委託する場合の取決め事項(4) セキュリティ管理に関する事項① データの入力、修正、削除等に関する担当者のアクセス権限の設定と不正アクセス防止に関する事項② 識別構成要素の管理に関する事項③ ハードウェア設置場所への立入制限に関する事項(5) バックアップ・リストアに関する事項(6) 変更の管理に関する事項① 変更の計画、承認の手順に関する事項② 変更の影響評価に関する事項③ その他、変更に必要な事項(7) 逸脱(システムトラブル)の管理に関する事項① 逸脱(システムトラブル)発生時の対応のための組織等② 逸脱(システムトラブル)の原因の究明及び影響評価③ 再発防止策等に関する事項④ 回復措置に関する手順⑤ システム停止後の再開手順及び再開時の確認事項⑥ その他逸脱の管理に必要な事項(8) 担当者の教育訓練に関する事項(9) 自己点検に関する事項 |
| 6.3 コンピュータ化システムの操作の手順に関する文書(標準操作手順書)の作成(1) 6.2(2)に規定する標準操作手順書には以下の事項を記載する① システムの担当者② コンピュータ化システムの操作に関する事項③ コンピュータ化システムの保守点検に関する事項④ コンピュータ化システムのセキュリティ管理に関する事項⑤ その他、コンピュータ化システムの特性に応じた運用管理に必要な事項(2)運用責任者は、標準操作手順書に基づき、担当者にコンピュータ化システムを操作させるものとする。 |
| 6.4 保守点検事項の実施運用責任者は、運用管理手順書に基づき、次に掲げる事項を行うものとする。(1) 担当者に保守点検を実施させ、その結果を記録させること。(2) 保守点検の記録により保守点検管理が適切に行われていることを確認すること。 |
| 6.5 セキュリティ管理の実施運用責任者は、運用管理手順書に基づき、次に掲げる事項を行うものとする。(1) データの入力、修正、削除等に関する担当者のアクセス権限の設定と、不正アクセスの防止措置を講じること。(2) 識別構成要素等の取扱いについて、機密保護を図ること。(3) 必要に応じてハードウェア設置場所への立入制限を行うこと。(4) セキュリティ管理に関する記録を作成するとともに、これを保管すること。 |
| 6.6 バックアップ及びリストア運用責任者は、あらかじめ指定した者に対し、運用管理手順書に基づき、次に掲げる業務を行わせること。(1) ソフトウェア及びデータのバックアップを行うこと。(2) 障害発生からの回復のためにソフトウェア及びデータのリストアを行うこと。(3) バックアップ及びリストアに関する記録を作成するとともに、これを保管すること。 |
| 6.7 変更の管理運用責任者は、あらかじめ指定した者に対し、運用管理手順書に基づき、次に掲げる業務を行わせること。(1) 変更がコンピュータ化システムに与える影響を評価し、評価の結果に基づき適切な措置を実施すること。なお評価の結果、バリデーションが必要と判断された場合は、リスクの程度に応じて「4. 開発業務」及び「5. 検証業務」に戻ってバリデーションを実施すること。(2) 変更に伴い発生する手順に関する文書の変更箇所を特定し、必要な改定を実施すること。(3) 変更内容の関係者への周知の方法を決定し、必要に応じて教育訓練を実施すること。(4) 変更の管理の記録を作成し、運用責任者の確認を得るとともに、変更の管理に関する責任者等の承認を得ること。GMP省令に係るシステムに関する変更の管理については、GMP省令における変更の管理の手順に従って運用すること。ただし、その場合も上記(1)から(4)の内容を含むこと。 |
| 6.8 逸脱(システムトラブル)の管理運用責任者は、あらかじめ指定した者に対し、運用管理手順書に基づき、次に掲げる業務を行わせること。(1) 発生した逸脱(システムトラブル)が製品の品質に及ぼす影響を評価し、速やかに適切な対応措置を講じるとともに、その原因を究明し、必要な再発防止措置を実施すること。(2) 逸脱(システムトラブル)発生後にコンピュータ化システムの運用を再開する場合には、復帰稼働が適切に行われていることを確認すること。(3) 逸脱(システムトラブル)の管理の記録を作成し、運用責任者の確認を得るとともに、逸脱の管理に関する責任者等の承認を得ること。GMP省令に係るシステムに関する逸脱の管理については、GMP省令における逸脱の管理の手順に従って運用することでよいが、その場合も上記(1)から(3)の内容を含むこと。 |
| 6.9 教育訓練 6.9.1 教育訓練計画の作成運用責任者は、運用管理手順書に基づき、あらかじめ指定した者に、コンピュータ化システムを使用した業務に従事する者に対する教育訓練計画を作成させること。なお、教育訓練についてはGQP省令、GMP省令における手順に従って運用することが望ましい。6.9.2 教育訓練の実施運用責任者は、教育訓練計画に基づき、あらかじめ指定した者に次に掲げる業務を行わせること。(1) コンピュータを使用した業務に従事する者に対して、コンピュータ化システムを使用した業務に関する教育訓練を計画的に実施し、その記録を作成すること。(2) 教育訓練の実施状況について運用責任者の確認を得るとともに、品質保証責任者又は製造管理者若しくは責任技術者に対して文書により報告すること。6.9.3 教育訓練の記録の保管運用責任者は教育訓練の実施の記録を保管すること。 |
7.自己点検
| 7 自己点検 7.1 自己点検の実施製造販売業者等は、手順書等に基づき、あらかじめ指定した者に次に掲げる業務を行わせること。なお、自己点検においては、GQP省令、GMP省令における手順に従って運用することが望ましい。(1) コンピュータ化システムがこのガイドラインに基づき、運用管理されていることを確認するために定期的に自己点検を実施すること。(2) 自己点検の結果について品質保証責任者又は製造管理者若しくは責任技術者に対して文書により報告すること。(3) 自己点検の結果の記録を作成し、これを保管すること。7.2 改善措置の実施製造販売業者等は、自己点検の結果に基づき、改善が必要な場合には、あらかじめ指定した者に所要の措置を講じさせ、その記録を作成し適切な者に報告させるとともに、これを保管させること。 |
8.コンピュータシステムの廃棄
| 8. コンピュータシステムの廃棄 8.1 コンピュータシステムの廃棄の計画に関する文書の作成製造販売業者等はコンピュータシステムの廃棄にあたっては、コンピュータシステムの種類や規模、カテゴリ等、必要に応じて、コンピュータシステムの廃棄に関する計画書(以下「廃棄計画書」という。)を作成すること。廃棄計画書には原則として以下の事項を記載するものとする。(1) 廃棄に関する責任体制と役割-組織-コンピュータシステムの廃棄の責任者(2) 廃棄対象とするコンピュータシステム(3) データの移行に関する事項(4) セキュリティに関する事項(5) コンピュータシステムの廃棄方法コンピュータシステムの種類や規模、用途等に応じて以下を参考にして適切に定めること-リスクアセスメント-前提条件-スケジュール-具体的な廃棄の方法・ハードウェア・ソフトウェア・データ・文書類(手順書、記録、契約書等)(6) 廃棄完了の判断基準 |
| 8.2 コンピュータシステムの廃棄記録の作成コンピュータシステムの廃棄の責任者は、廃棄計画書に基づきコンピュータシステムを廃棄するとともに、廃棄の記録を作成し、これを保管すること。 |
9.文書及び記録の管理
| 9. 文書及び記録の管理このガイドラインに基づき作成された文書及び記録はGQP省令又はGMP省令に基づき定めた文書及び記録の管理の方法に従って適切に保存管理するものとする。GQP省令及びGMP省令にまたがるシステムの場合は、あらかじめどちらの省令に従って管理するかを運用管理手順書に明記しておくこと。 |
10.用語集
| 10. 用語集 運転時適格性評価(OQ)コンピュータ化システムが、稼働時と同等の環境下において、機能仕様に示された運転範囲で意図したように機能と性能を発揮することを確認し文書化すること。運用管理業務コンピュータ化システムの稼働開始後、コンピュータ化システムを、バリデートされた状態を維持し、要求仕様に記載された要件に基づいて適正に稼働させるための業務。運用責任者コンピュータ化システム運用業務を行うための責任者として、製造販売業者等により運用管理手順書において指定された者をいう。開発業務指定されたコンピュータ化システムの計画、設計、製作、テスト、受入試験までの業務。開発計画書指定されたコンピュータ化システムを開発する際に目的、条件、責任、体制、スケジュールなどを記述した文書。開発責任者コンピュータ化システム開発業務を行うための責任者として、製造販売業者等により開発計画書においてあらかじめ指定された者をいう。機能仕様書(FS)要求仕様書に記載された要求仕様に対応する、より具体的な機能が記載された文書。供給者コンピュータ化システムを開発あるいは導入し、製造販売業者等に提供する者をいう。一般にサプライヤやベンダーと呼ばれる。自社で開発する場合は自社のシステム開発者も含む。供給者アセスメント製造販売業者等による供給者の選定時から開発の各段階における供給者の評価のこと。供給者監査供給者の品質管理体制や品質保証のシステム、あるいは経験・能力や実績など多角的に供給者の調査を行い、供給者の総合的な品質マネジメントシステムや能力を評価・確認すること。実地又は書面による監査方法がある。検証業務コンピュータ化システムが、要求仕様等に定めた要件に合致して設計され、据え付けられ、システムの稼働環境及び稼働状態において、機能及び性能を発揮することを確認すること。検証責任者検証業務を行うための責任者として、製造販売業者等により開発計画書においてあらかじめ指定された者をいう。現地受入試験(SAT, Site Acceptance Test)供給者がシステムを現地の稼働環境で機能及び性能の全てあるいは一部が機能仕様を満足していることを確認すること。ここでいう「現地」とは、製造販売業者等が当該のシステムを設置する予定の場所をいう。 工場出荷試験(FAT, Factory Acceptance Test)供給者がシステムを出荷する前に製作環境で機能及び性能の全てあるいは一部が機能仕様を満足していることを確認すること。構成設定コンピュータシステムを利用するにあたってハードウェア及びソフトウェアの構成要素の組み合わせや稼働条件等を設定すること。すなわち、ハードウェアにおいては、システムを構成する、コンピュータ、周辺機器あるいはそれらに組み込まれる部品(ボード等)の組み合わせを設定し、登録すること。ソフトウェアにおいては、プログラムを作成、変更することなく、システムを構成するモジュールの組み合わせ、及びシステムが稼働する条件、パラメータ等を設定し、登録すること。コンピュータシステム特定の機能又は一連の機能を実行するために、設計し、組み立てられたハードウェア及び関連するソフトウェアのグループ。コンピュータ化システムコンピュータシステムで統合された工程又は作業(原薬GMP)、及びコンピュータシステムにより実現される機能を利用する業務プロセス。コンピュータシステムの廃棄の責任者コンピュータシステムの廃棄を行うための責任者として、製造販売業者等により廃棄計画書において指定された者をいう。識別構成要素システムの運用において操作者を識別、特定するために用いられるデータの組み合わせ、もしくは機器とデータの組み合わせ、例えばIDとパスワードの組合せ。システムアセスメント開発対象とするコンピュータ化システムのバリデーションにおける検証内容や作成文書等を決定するために、システムのソフトウェアの複雑性や開発方法、当該システムにより製造される製品の安全性や品質への影響の度合い、又は当該システムにより作成、保存される電子記録の重要度、供給者のシステム開発過程での品質保証の状況等を総合的に評価すること。システムテスト稼働するために結合された状態でモジュール、プログラムが機能仕様書、設計仕様書通りに機能することを確認すること。据付時適格性評価(IQ)システムが、要求仕様等に記載されたとおりに据え付けられ、プログラムがインストールされたことを確認し、文書化すること。性能適格性評価(PQ)コンピュータ化システムが、稼働時において、要求仕様通りに機能し、性能を発揮して運転できることを確認し、文書化することをいう。設計仕様書(DS)機能仕様に記載された具体的な機能を実現するコンピュータ化システムを作成するための詳細仕様が記載された文書で、ハードウェア仕様書とソフトウェア仕様書に分けられる場合がある。ハードウェア仕様書:システムを構成するハードウェアの仕様、構成を記述するソフトウェア仕様書:システムを構成するソフトウェアの詳細機能、構成を記述する設計時適格性評価(DQ)要求仕様書に記載された要求事項が、以降の機能仕様書、設計仕様書に正しく反映されていることを確認し文書化すること。ソフトウェアカテゴリ同程度の信頼性を有するソフトウェアの属すべき範囲。ソフトウェアの性質や特徴を区分する上での基本的な分類。プログラム仕様書設計仕様書の機能を実現するためにモジュール、プログラムで実現すべき事項を記述した仕様書。プログラムテストモジュール、プログラムが単体でプログラム仕様書通りに機能することを確認すること。モジュールソフトウェアを構成する機能の最小単位。要求仕様書(URS)指定されたコンピュータ化システムに関する機能上の要求仕様が記載された文書。リスクアセスメントリスクマネジメントプロセスの中で、リスクに係わる決定を支持する情報を整理する系統だったプロセス。ハザードの特定、及びそれらハザードへの曝露に伴うリスクの分析と評価からなる。リストアあらかじめ適切な媒体にバックアップしておいた、プログラム、パラメータ、データ等を、再度システムに読み込ませ、システムをバックアップした時点と同様の状態に戻すこと。その他の用語については、GQP省令、GMP省令及び関連の通知類の用語を参照。 |
別紙1 コンピュータ化システムのライフサイクルモデル
別紙2 カテゴリ分類表
参考
- 医薬品・医薬部外品製造販売業者等におけるコン ピュータ化システム適正管理ガイドライン(案) 厚生労働省 2010年7月16日








