
構造設備のCSV実施方法
2026年最新動向アップデート(構造設備のCSV)
本記事は構造設備のCSVを扱ったものです。コンピュータ化システムバリデーション(CSV)の周辺規制は2022〜2026年に大きく動きました。
主要規制・ガイダンスの最新化
- ISPE GAMP 5 Second Edition(2022年7月):CSA、AI/ML(Appendix D11)、クラウド、ブロックチェーン、OSS、ALCOA+ Data Integrityを大幅追加。第1版(2008)からの大改訂。
- FDA CSA最終ガイダンス(2025年9月/2026年2月3日改訂版でQMSR整合):文書中心のCSVから、リスクベース・継続的アシュアランスへ。
- FDA QMSR施行(2026年2月2日):21 CFR Part 820がISO 13485:2016を引用組み込み。
- 21 CFR Part 11はData Integrity(ALCOA+)と組み合わせた運用が標準化。
- EU GMP Annex 11とPIC/S Annex 11も改訂作業中。
実務への含意
Data Integrity(ALCOA+)+ CSA(リスクベース検証)+ QMSR(ISO 13485:2016 + FDA追加要件)+ GAMP 5 2nd Editionの四位一体で運用設計するのが、2026年以降の標準です。
※以下は本記事のオリジナル解説です。
コンピュータ化システムの4つの種類
コンピュータ化システムは、大きく分類して4種類のカテゴリに分けられる。(図1参照)
1) プロセスコントロール(構造設備)
2) ITアプリケーション
3) ラボ(分析機器、Excel)
4) インフラストラクチャ
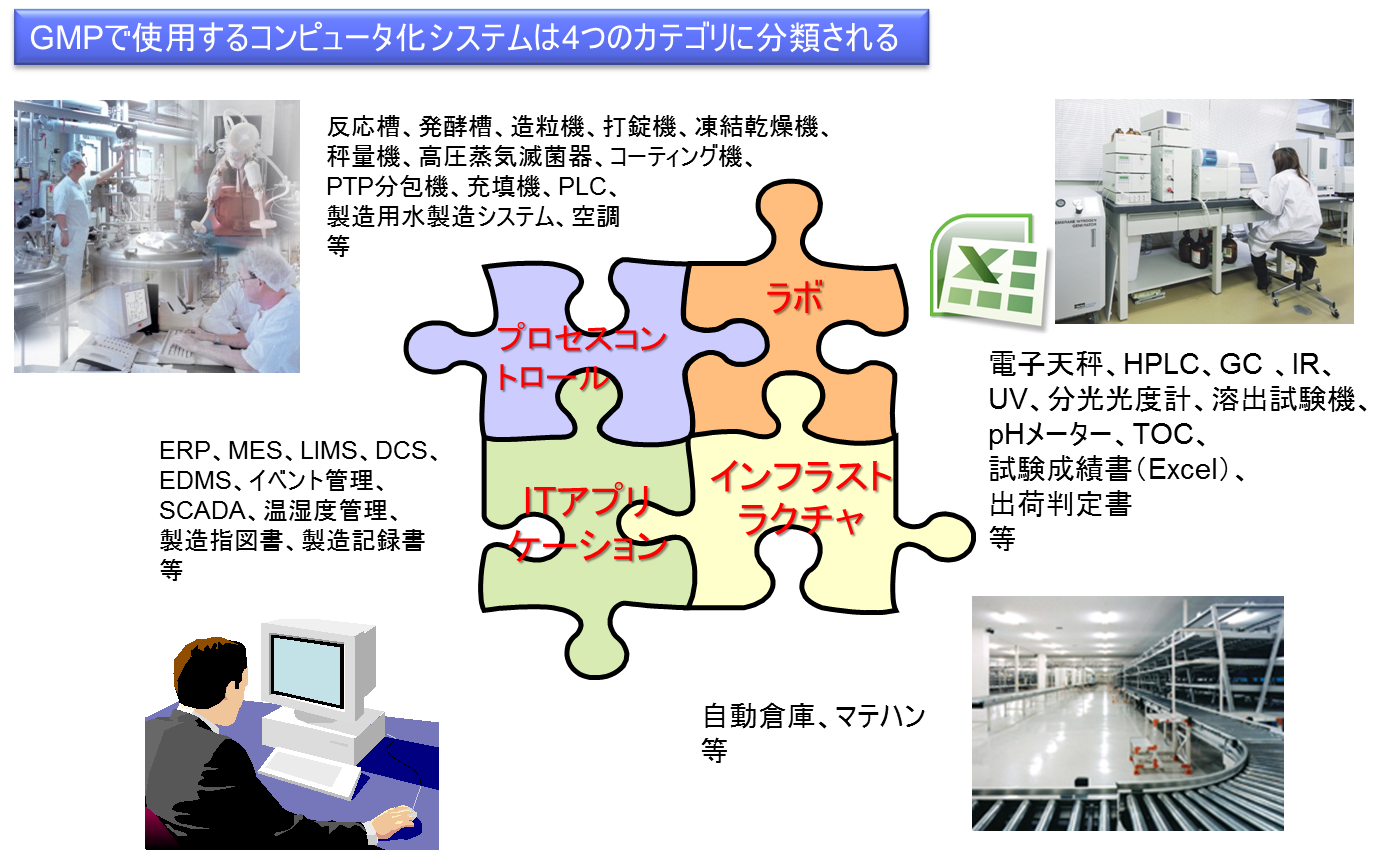
一般に、GMP関連業務においては、上記の4システムがすべて使用される。それに対して、GLP関連業務では、主にラボとITアプリケーションが使用され、GCP・GVP・GQP関連業務では、ITアプリケーションが使用される。
上記4種類のシステムは、それぞれに特徴が異なり、またバリデーションの実施方法が異なる。しかしながら、どのカテゴリにも精通した専門家はほとんどいないのが現状である。
プロセスコントロール(構造設備)
構造設備は、工場のラインに設置され、実際に原薬や製品(製剤)を生産するシステムである。
例えば、原薬工場における反応槽、発酵槽や、製剤工場における造粒機、打錠機、凍結乾燥機、秤量機、高圧蒸気滅菌器、コーティング機、PTP分包機、充填機などが相当する。
また、製造用水製造システムや空調などの支援設備(ユーティリティシステム)も構造設備に含まれる。
構造設備の品質は、製品の品質に大きく影響する。したがって、バリデーションは重要である。
「原薬GMPのガイドライン」では、プロセスバリデーションを始める前に、重要な装置及び付帯設備の適格性評価を完了することとなっている。
構造設備の特徴は、その品質が直感的に把握できることにある。すなわち、あらかじめ実際に製品を生産し、生産された製品の品質を目視チェックや分析することによって検証ができるのである。
構造設備では、PLC、ファームウェアなどの比較的小さなプログラムで制御していることが多い。
多くの構造設備は、カテゴリ3である。ただし、複雑またはユーザが変更したPLCは、カテゴリ5に分類されるが、カテゴリ3と5の境界は曖昧である。
一般に1つの構造設備は、1つの機能しかもたない。例えば、造粒機は造粒する機能、打錠機は打錠する機能である。したがって、多くの場合、構造設備では、機能仕様書は作成しない。(というよりも作成できない。)
構造設備を対象とするバリデーションを「適格性評価」と呼び、DQ(設計時適格性評価)、IQ(設備据付時適格性評価)、OQ(運転時適格性評価)、PQ(性能適格性評価)から構成される。
適格性評価は、製品品質に直接影響する要因についてのみ、設計段階でDQを、製作・施工段階でIQを、試験・検査・試運転段階でOQとPQを行うことである。
昨今は、SCADAやDCSのように、ネットワーク化され、ITアプリケーションにより中央で集中管理されている構造設備も利用されている。
旧ガイドラインではファームウェアやPLCは対象となっていなかった
平成4年2月21日に厚生省(当時)から発出された、薬監第11号「コンピュータ使用医薬品等製造所適正管理ガイドライン」(以下、旧ガイドライン)の「第2.適用範囲」には、以下の記載があった。
このガイドラインは、医薬品GMPが適用される製造所のうち、次のいずれかに該当するシステムを使用する製造所に適用する。
ただし、使用目的が限定され、そのためのプログラムがハードウェア(コンピュータにより制御される機器及び設備を含む。以下同じ。)の提供業者によって汎用機能として固定され、パラメーターを設定することによって機能が実現されるシステムを除くものとする。
この記述を読むと、旧ガイドラインでは、打錠機などの出来合いの構造設備(カテゴリ3)は除外されていたことがわかる。またファームウェアやPLC なども適用されていなかった。
新ガイドラインでは、カテゴリ3 に分類される構造設備や、それらに搭載されているPLC、ファームウェアも適用対象になったことに留意する必要がある。
図に示すとおり、新ガイドラインの別紙2にカテゴリ分類表が添付されている。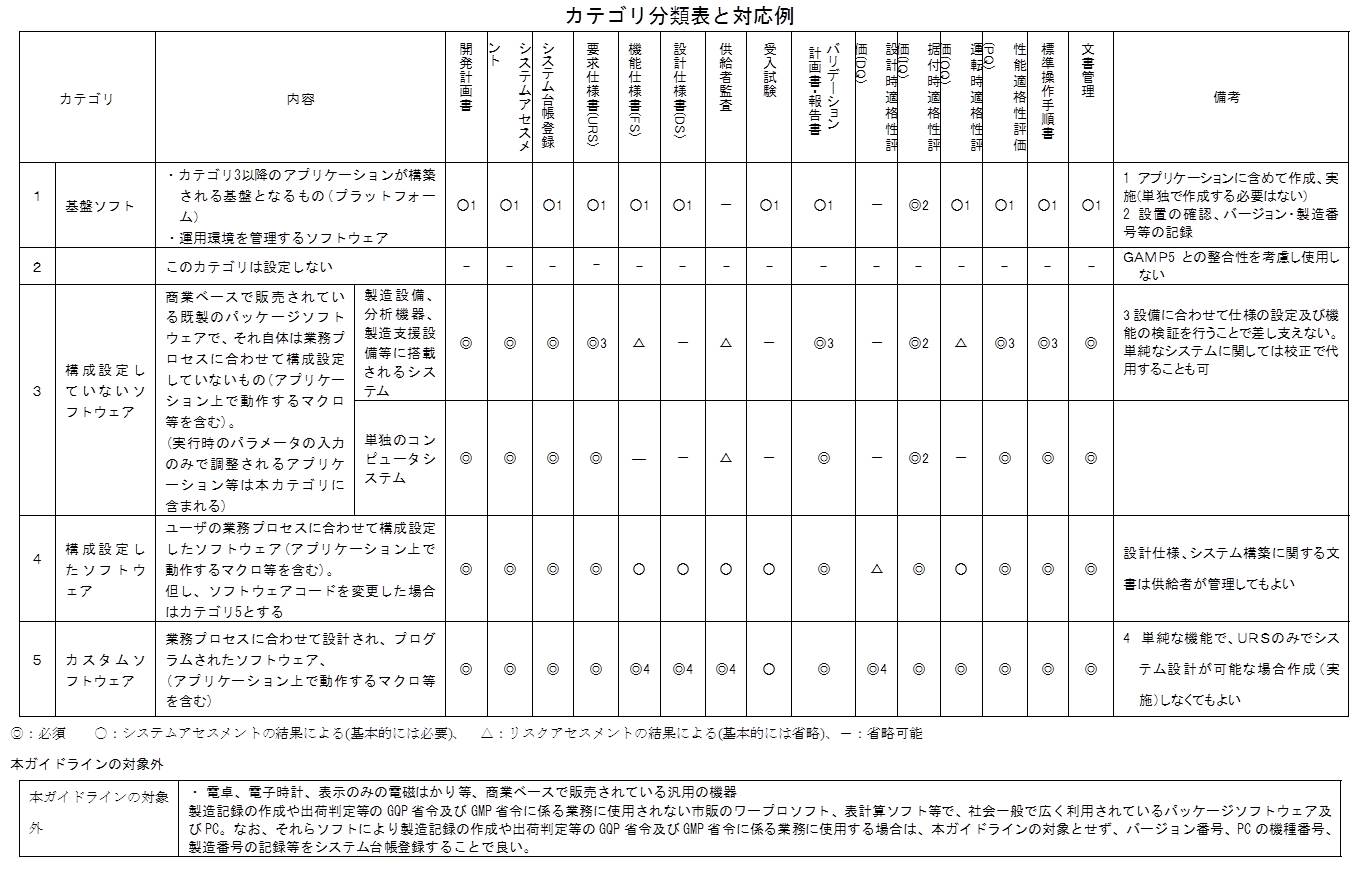
カテゴリ2は使用しないと明記されているが、これがGAMP 4では、ファームウェアやPLCであった。
実は、ファームウェアやPLCはCSV対象から外れたのではなくて、カテゴリ3の上段に移動された。
ここには「製造設備、分析機器、製造支援システム等に搭載されるシステム」とある。例えば、シーケンサー等がそれに相当するが、ファームウェアやPLCによって動作している。
GMPにおけるハードとソフト
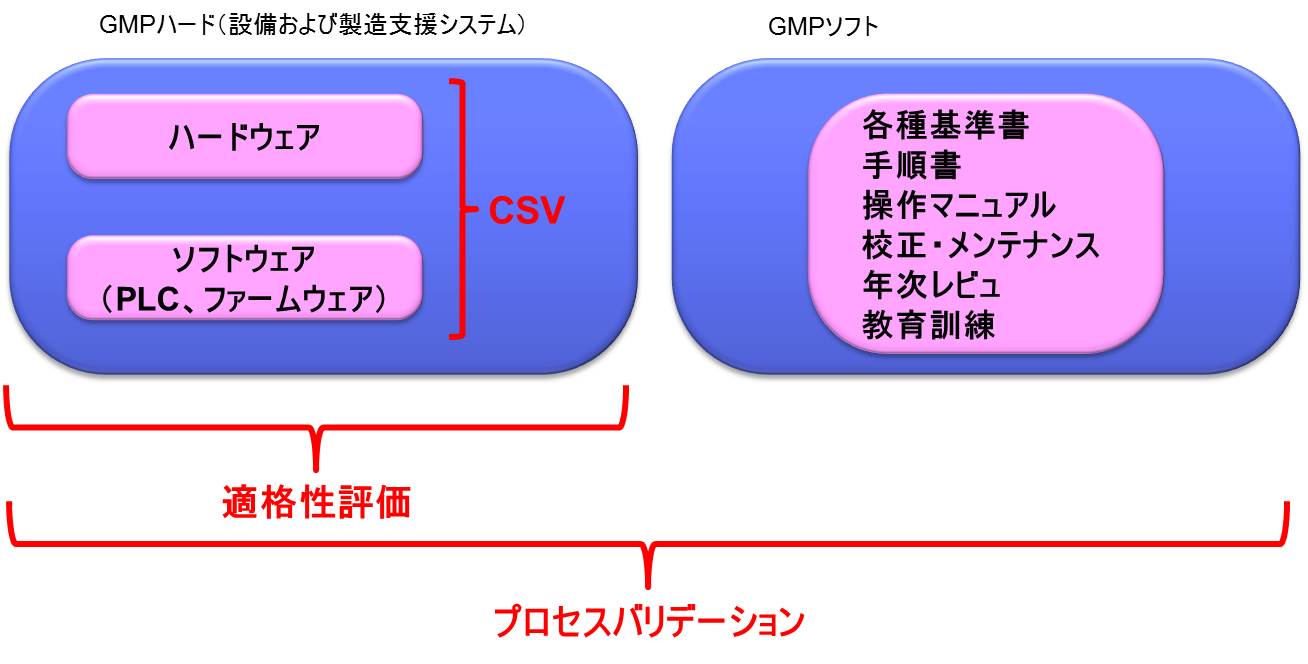
GMPを語る上でははずせない要素・考え方として「ハード」と「ソフト」がある。
「GMPハード」と「GMPソフト」の両者によりGMPの目的を達成するのである。
GMPハードとは、設備のことであり、例えば以下のような要件である。
- 間違いを防ぐことのできる設備・環境の製造所であること
- 衛生的な設備・環境の製造所であること
- 高い品質を保ち続けることができる設備・環境の製造所であること
一方、GMPソフトとは、ルールのことであり、以下のような要件があげられる。
- ルールを決めて文書化すること
- ルールどおりに実施し、記録を作成すること
- 定期的に見直しを行い、改善をはかること
ここで、「GMPハード」と「GMPソフト」は、「ハードウェア」、「ソフトウェア」のことではないことに注意が必要である。「ハードウェア」、「ソフトウェア」はともに「GMPハード」である。
適格性評価とは
GMPにおいて、「適格性評価」(Qualification)と呼ばれる、他の業界では使用されない特殊な用語が存在する。一般に構造設備のようなGMPハードのバリデーションについては、適格性評価を行う。
では、「適格性評価」とは、いったいどのようなものであるのだろうか。
ICH Q7を受けて平成13年に発出された「原薬GMPのガイドライン」(平成13年11月2日、薬発第1200号)の12.30において、以下のような記載がある。
12.30 プロセスバリデーションの作業を始める前に、重要な装置及び付帯設備の適格性評価を完了すること。適格性評価は、通常、以下の作業を個々に、又は組み合わせて実施する:
-設計時適格性評価(DQ):設備、装置又はシステムが目的とする用途に適切であることを確認し文書化すること。
-設備据付時適格性評価(IQ):据付け又は改良した装置又はシステムが承認を受けた設計及び製造業者の要求と整合することを確認し文書化すること。
-運転時適格性評価(OQ):据付け又は改良した装置又はシステムが予期した運転範囲で意図したように作動することを確認し文書化すること。
-性能適格性評価(PQ):設備及びそれに付随する補助装置及びシステムが、承認された製造方法及び規格に基づき、効果的かつ再現性よく機能できることを確認し文書化すること。
「プロセスバリデーションの作業を始める前に、重要な装置および付帯設備の適格性評価を完了すること」と記載のとおり、適格性評価はプロセスバリデーションを実施する前提条件であることがわかる。
わかりやすく説明をすると、「プロセスバリデーション」は、「GMPソフト」と「GMPハード」の両方に対して実施するのに対し、「適格性評価」は「GMPハード」に対して実施する。
適格性評価は、DQ(設計時適格性評価)、IQ(設備据付時適格性評価)、OQ(運転時適格性評価)、PQ(性能適格性評価)から構成される。
FDAが2002年1月11日に発行した「General Principles of Software Validation; Final Guidance forIndustry and FDA Staff」の「3.1.3 IQ/OQ/PQ」には、以下の記載がある。
長期にわたり、FDAと規制の適用を受ける業界は、プロセスバリデーションにおける専門用語で、ソフトウェアバリデーションの理解や定義付けを試みた。例えば、業界文書やその他FDAバリデーションガイダンスは、installation qualification (IQ:設置適格性検証)、 operational qualification (OQ:稼動適格性検証) 、performance qualification (PQ:性能適格性検証)の観点からユーザによるソフトウェアバリデーションを何度か記載している。
(中略)
IQ、OQ、PQの専門用語はその目的に十分沿い、ユーザ側でのソフトウェアバリデーションタスクを系統づける、数ある合法的な方法の一つではあるが、この専門用語は多くのソフトウェア専門家の間ではよく理解がされていないおそれがあり、本ドキュメントでも別の箇所では扱っていない。しかしながら、FDA職員と機器製造業者は、ソフトウェアバリデーションに関する情報を求め、提供する立場にあることから、これら用語の違いを把握することが必要となる。
すなわち、CSVで使用するIQ、OQ、PQという用語は、プロセスバリデーションの用語を流用したものであることがわかる。
しかし、一般的にIQ、OQ、PQという用語は、製薬業界の特殊用語で、いわば方言である。したがって、一般のITベンダー等が理解できるとは限らないことをFDAは懸念している。
また「原薬GMPのガイドライン」の用語集に、以下のような定義が記載されている。
バリデーション
特定の工程、方法又はシステムが、一貫して、予め設定した判定基準に適合する結果を与えるという高度の保証を提供する文書によるプログラム。
適格性評価
装置又は付帯システムが適切に据え付けられ、正しく作動し、実際に期待される結果が得られることを証明し、記録する活動。適格性評価はバリデーションの一部であるが、個々の適格性評価のステップのみではプロセスバリデーションとはならない。
つまり、バリデーションの活動の中に、適格性評価が含まれていることがわかる。
CSVとプロセスバリデーション(PV)の違い
かつては構造設備は、電動であったとしても人が制御していた。
しかしながら、昨今の構造設備は、コンピュータによって制御されることがもっぱらとなった。
このように人ではなくコンピュータによって制御されたシステムのことを「コンピュータ化システム」と呼ぶ。
構造設備に搭載されているコンピュータの多くは、ファームウェアやPLCといった比較的小さなプログラムで構成されていることが多い。
したがって、CSVはファームウェアやPLCといったプログラムの品質保証が中心となるのである。
この理解をすれば、CSVとPVの違いは明白となる。
医薬品の生産プロセスを保証するためには、まず構造設備に搭載されているシステム(ファームウェア、PLC等)の品質保証を行うことが重要であり、これがCSVの目的である。
つまり、コンピュータによって制御された構造設備(コンピュータ化システム)の適格性評価は、CSVを実施することに他ならない。
逆に言うと、コンピュータ化されていない構造設備は、単に適格性評価を行うこととなる。
CSV(適格性評価)によって品質が保証された構造設備-つまりGMPハードについて、手順書等のGMPソフトを合わせてプロセスバリデーションを実施することとなるのである。
多くのセミナーや書籍では、CSVとPVを混同して解説しているものが多いので、注意が必要である。
このことにより、製薬企業の担当者の多くが、CSVとPVの違いが全く分からなくなってしまっているように思われる。
カテゴリ2について
前述した通り、旧ガイドラインではファームウェアやPLCは、CSVの対象とされていなかった。
しかしながら、欧州とりわけイギリスでは従来からファームウェアやPLCのバリデーションには厳しかった。一方でFDAは、それらについてCSVの要否を明らかにして来なかった。そこでGAMP 5では、従来からカテゴリ2に分類されてきたファームウェアをカテゴリ3に含めることとなった。
すなわち、ファームウェアやPLCは、CSVの対象となったのである。
国際整合をとるため、新ガイドラインでは、やはり対象となった。
ファームウェアは、構造設備や分析機器にも使用されている。
新ガイドラインは、平成22年10月に発出されたが、施行まで1年半という異例の長さの移行期間が設けられた。その理由は、旧ガイドラインでは対象となっていなかったコンピュータ化システムの回顧的バリデーションの実施であったものと理解している。
しかしながら、いったいファームウェアやPLCを搭載した構造設備のCSVはどのように実施するべきなのであろうか。
ちまたでは、ベンダーに構造設備のCSVを依頼したところ、高額な見積書を受け取ったという話をよく耳にする。
ベンダーも過剰にCSVを考えてしまっているきらいがある。
新ガイドラインにおける構造設備のCSV実施方法
新ガイドラインの別紙2「カテゴリ分類表」では、カテゴリ2は使用しないと明記されているが、これがGAMP 4では、ファームウェアやPLCであった。
実は、ファームウェアやPLCはCSV対象から外れたのではなくて、カテゴリ3の上段に移動された。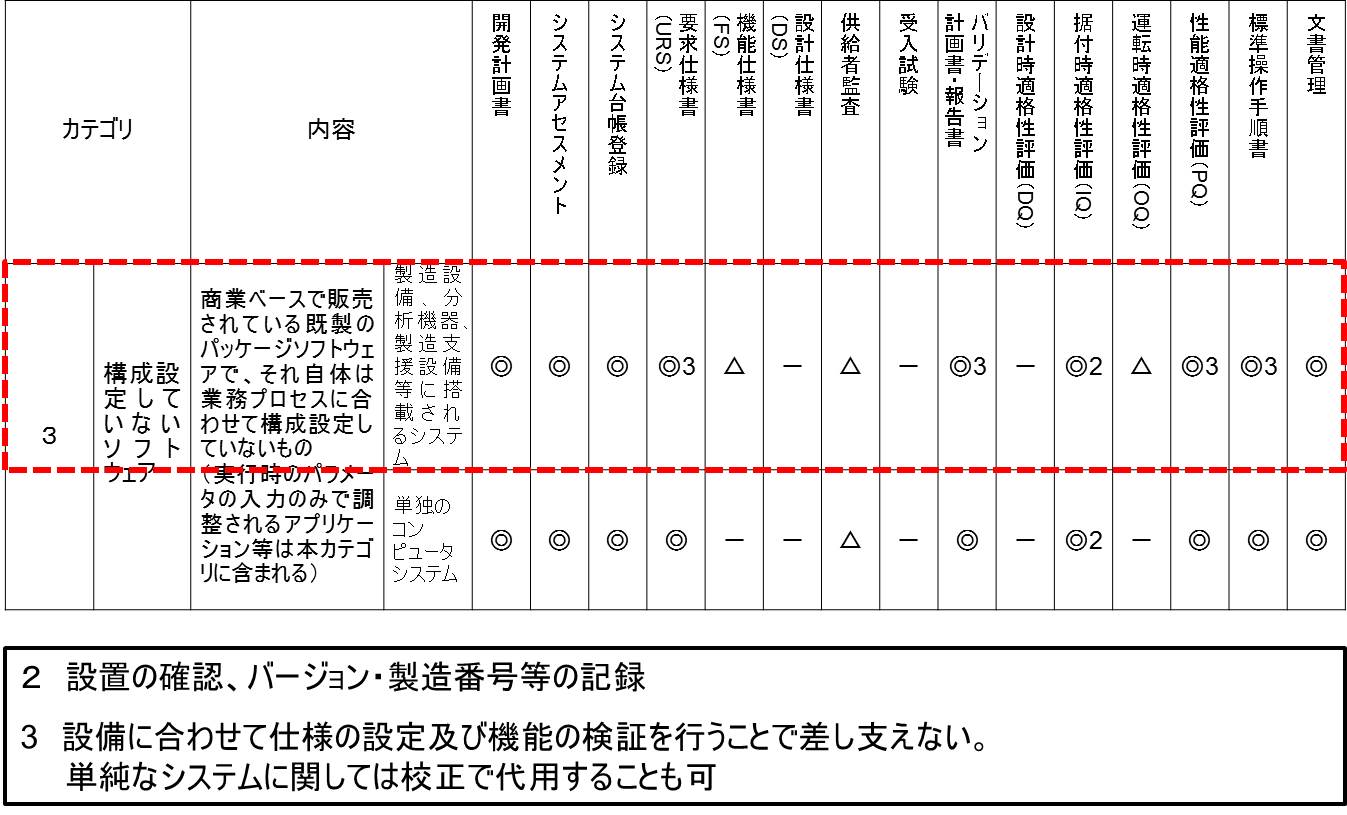
ここには「製造設備、分析機器、製造支援システム等に搭載されるシステム」とある。例えば、シーケンサー等がそれに相当するが、ファームウェアやPLCによって動作している。
PLCは、カテゴリ分類が難しい。一般にPLCはカテゴリ3に分類されるが、複雑なものやカスタムラダーロジックなどはカテゴリ5に分類される。その境界線はグレーである。
カテゴリ分類表のガテゴリ3の上段を詳細に考察してみたい。
まず、開発計画書は◎であるので作成は必須である。
システムアセスメント-つまりカテゴリ分類・品質リスクアセスメント・供給者アセスメント-も◎であり必須である。
当然のことながら、システム台帳登録も◎であり必須である。
要求仕様書(URS)も、◎であり必須である。
開発計画書、システムアセスメント、要求仕様書は合わせて1冊で執筆した方が効率が良いと思われる。
機能仕様書(FS)は△と記載されているが、凡例には「基本的には省略」とある。したがって、通常は作成しない。というよりも作成できない。
設計仕様書(DS)は、-であり、作成不要である。
供給者監査は、△であるので、基本的には省略する。
受入試験は、-であり、実施不要である。
バリデーション計画書・報告書は◎であるので、作成は必須である。ただし、ルビに3と振ってあり、備考欄には「3 設備に合わせて仕様の設定及び機能の検証を行うことで差し支えない。」と記載されている。これは包装設備などにおいて、複数台のシーケンサーが搭載されている場合、個別にバリデーションを行うのではなく、設備全体として検証しても構わないことを意図していると思われる。
設計時適格性評価(DQ)は、-であり、実施しない。
据付時適格性評価(IQ)は、◎になってはいるが、ルビに2と振ってあり、備考欄には「2 設置の確認、バージョン・製造番号等の記録」とある。つまり、IQとしての作業は事実上、実施しないことになる。
運転時適格性評価(OQ)は、△であるので、基本的には省略する。つまり、機能仕様書を作成しないので、OQは実施できない。
性能適格性評価(PQ)は、◎ではあるが、バリデーション計画書・報告書と同様に3とルビが振ってある。
構造設備の場合、事実上PQはプロセスバリデーションによって検証することになる。したがって、CSVにおけるPQは省略できるのである。
また、分析機器のCSVにおけるPQ実施は、備考欄に記載がある通り、「単純なシステムに関しては校正で代用することも可」である。つまり、分析機器(インテグレータが接続されているような複雑なものを除く)のPQでは、校正を実施するのみとなり、事実上PQを省略できる。
標準操作手順書(SOP)は、作成が必須である。しばしば標準操作手順書を作成せずに、サプライヤから提供された操作マニュアルをそのまま使用しているケースを見かけるが、これは避けなければならない。
なぜならば、操作マニュアルには、実際の作業では使用しない操作が記載されていることがあり、作業員が間違った操作を行ってしまう根本原因となるからである。
EU GMP Annex 11の3.3には、以下のような要求がある。
既成のソフトウェアで提供された文書は、規制関係ユーザによってレビュされ、ユーザ要件を満たしているかチェックされなければならない。
基本的には、標準操作手順書の作成は、必須であることを理解するべきである。
最後に文書管理は、◎であり、必須である。あらゆるCSV成果物は、きちんと整理された状態で保存しておき、監査や査察時などに適切に提示できるようになっていなければならない。








