FDA cGMPが改定されない理由
「古い本体と新しい補遺」のモデル
基本構造の固定と柔軟な補完
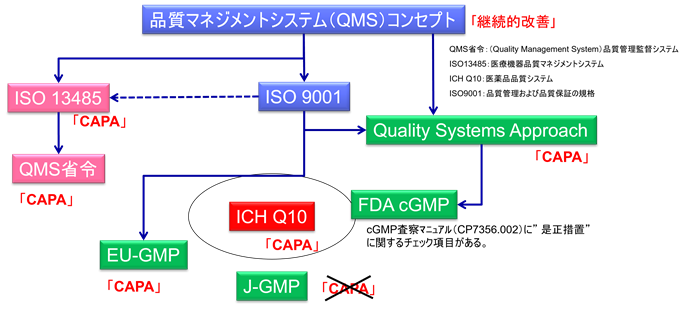
FDA cGMPの本体である連邦規則集(21 CFR 210/211)は、1963年の初版以降、根本的な改定がほとんど行われていない。これは単に改定が怠られているわけではなく、FDAの規制アプローチに起因している。基本的な要件は法的拘束力のある規則として維持しつつ、新たな科学的知見や技術的進歩に対応するためにガイドラインという形で補完するという二層構造を採用しているのである。
これは日本のGMP省令と課長通知の関係に類似している。日本においても、GMP省令が基本要件を定め、より詳細な運用や解釈は課長通知などの行政文書によって補完されている。
見えにくくなる規制要件の全体像
このアプローチの課題は、「何が法的に要求されているのか」の全体像が見えにくくなることである。例えば、CAPA(是正措置・予防措置)について考えてみよう。
FDA cGMPの本体には、直接的なCAPA要求は明記されていない。これは本体が制定された1960年代には、現代的な品質システムの概念が十分に発達していなかったためである。しかし、製薬業界の発展と共に品質システムの重要性が認識され、FDAは2006年に「Guidance for Industry: Quality Systems Approach to Pharmaceutical CGMP Regulations(品質システムアプローチ)」というガイドラインを発行した。このガイドラインでは、cGMPを現代的な品質システムの文脈で解釈し、CAPAを含む品質マネジメントシステムの要素を導入している。
このように、法的拘束力を持つ規則本体には明記されていないが、ガイドラインを通じて事実上要求されている事項が数多く存在する。製薬企業は、規則本体だけでなく、関連する全てのガイドラインを把握し、統合的に理解する必要がある。
PIC/S GMP:「統合型」モデル
対照的に、EU GMPやPIC/S GMPは比較的シンプルな構造を採用している。これらは本体と附属書(Annexes)から構成され、規制要件が一つの文書体系内に統合されている。
この統合型アプローチの利点は、規制要件の全体像が把握しやすいことである。例えば、品質システムに関する要件はPart I Chapter 1に明確に記載されており、CAPAの要求も明示的に含まれている。また、附属書も本体と同等の位置づけで、特定の分野や活動に関する追加要件を定めている。
この構造により、EU GMPやPIC/S GMPは、FDA cGMPと比較して規制要件の透明性が高く、製薬企業にとっては遵守すべき事項の特定がより容易になっている。

PIC/S GMP 本体(Part1)

PIC/S GMP Annexes
規制アプローチの違いが生み出す国際調和の課題
FDA cGMPとEU GMPの構造の違いは、国際的な規制調和にも影響を与えている。PIC/S(医薬品査察協同スキーム)を通じた国際的な調和の取り組みがあるものの、基本的な規制アプローチの違いが完全な調和を難しくしている。
日本は2014年にPIC/Sに加盟し、GMP省令もPIC/S GMPに準拠するよう改正された。これにより、日本のGMP規制はEU型の統合モデルに近づいてきたが、FDA cGMPとの差異は依然として存在する。
製薬企業にとっての実務的示唆
このような規制構造の違いを理解することは、グローバルに事業を展開する製薬企業にとって重要である。特に、以下の点に注意が必要である:
FDA市場向け製品の製造: FDA cGMPに準拠するためには、連邦規則集だけでなく、関連する全てのガイドラインも把握し、統合的に理解・適用する必要がある。
EU市場向け製品の製造: EU GMPの本体と附属書の要件を把握し、適用する。
グローバル品質システムの構築: 各地域の規制要件の違いを考慮しつつ、最も厳格な要件を満たすグローバル品質システムを構築することが効率的である。
規制情報の継続的モニタリング: 特にFDAの場合、新たなガイドラインの発行や既存ガイドラインの改定が頻繁に行われるため、継続的なモニタリングが必要である。
関連商品