第4章 QMSRの要点
4.1 QMSRの要求事項の基本的な考え方
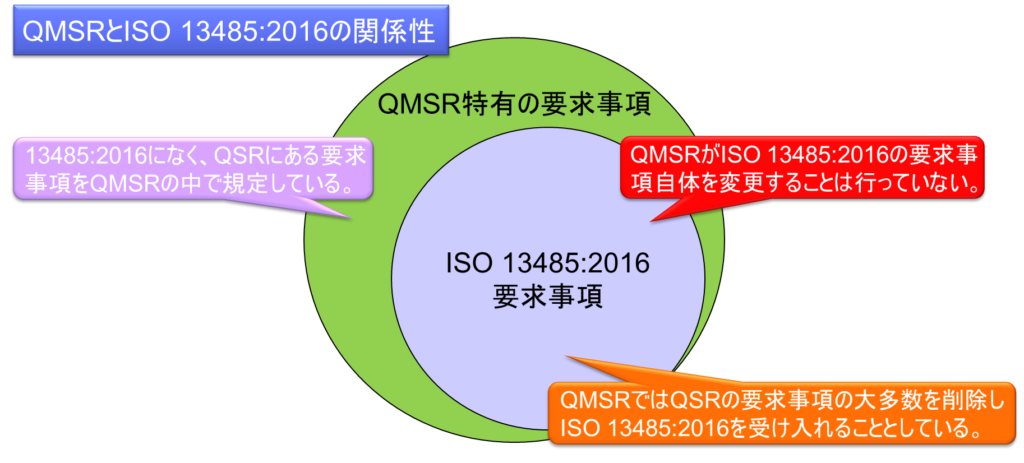
QMSRは、従来のQS規則(品質システム規制)の要求事項の大多数を削除し、ISO 13485:2016を全面的に取り入れるという革新的なアプローチを採用している。この戦略的な規制改正は、医療機器の品質管理システムに関する国際的な調和と標準化を実現するための重要な取り組みである。
具体的には、QMSRは以下の基本的な戦略を採用している:
まず、ISO 13485:2016 にはなく、QS 規則に存在していた要求事項を QMSR の中で規定しています。これにより、重要な規制要件が完全に失われることを防いでいます。
第二に、ISO 13485:2016の要求事項自体を変更するために、FDA特有の追加要求事項を組み込むアプローチを選択している。この方法により、国際標準との整合性を維持しつつ、米国固有の規制ニーズにも対応することができる。
4.2 QMSRの要求事項の実質的同等性:要点1.QMSRの要求事項は実質的にQSRとほぼ同じである
QMSRの要求事項は、現行のQS規則と実質的に同等であると詳細に評価している。 この評価の根拠は、現行のPart 820(QS規則)の要求事項が、全体としてISO 13485:2016の要求事項と非常に似ており、QS規則に特有の要求事項がQMSRにおいても維持されていることにある。
この実質的な同等性は、医療機器製造業者にとって非常に重要な意味を持つ。

4.3 ISO 13485:2016の参照による組み込み:要点2.ISO 13485:2016をそのまま引用し、追加事項要求事項を加えたもの
QMSRは、ISO 13485:2016をそのまま引用し、追加の要求事項を引き続き独自のアプローチを採用している。この方法には重要な特徴がある:
まず、QMSR に準拠したから推奨、自動的に ISO 13485:2016 の認証を取得できるわけではありません。逆に、ISO 13485:2016 の認証を持っていることが、QMSR への適合を保証するわけない。
この戦略により、FDAは医療機器の品質マネジメントシステムに関する国際的な要求事項を取り入れつつ、米国固有の規制要件を維持することができる。

4.4 QMSRの遵守と国際規格の関係:要点3.QMSRの遵守=ISO 13485:2016の遵守に繋がるように設計されている
QMSRは、ISO 13485:2016の要求事項を遵守することが、同時にQMSRの要求事項を準拠することにつながるように設計されています。 ただし、重要な点として、FDA独自の特定の要求事項も併せて遵守する必要がある。
QMSR の要求事項の大部分は、ISO 13485:2016 を受け入れることで対応される。の中で明確に規定されている。
これにより、国際標準との整合性を考慮しながら、米国独自の規制要件も確実に遵守されることになる。
4.5 国内規制との比較
日本のQMS契約と米国のQMSRの立て付けには、重要な構造的な違いが存在する。これに対し、米国のQMSRは、820.7からISO 13485:2016を参照させて規則的に取り込み、米国特有の要求事項をQMSR本文に追加的に規定している点が異なる。
具体的には、米国のQMSRは以下の特定の要求事項を追加している:
- 820.1:適用範囲
- 820.3:用語の定義
- 820.7:参照による組み込み
- 820.10:品質マネジメントシステムの要求事項
- 820.35:記録の管理
- 820.45:機器のラベリングおよび梱包の管理

4.6 追加の要求事項の詳細
QMSRには、ISO 13485に対する重要な追加の要求事項が存在します。これらの追加要求事項は、主に以下の領域に焦点を当てている:
- 記録の管理に関する要求事項 FDA は、ISO 13485:2016 に含まれていない特定の記録管理に関する要求事項を追加している。
- 苦情に関する詳細な記録要件
- 付属帯サービスに関するその他の記録
- 医療機器固有識別子(UDI)に関する記録
- コンフィデンシャルな記録の取り扱いに関する特別な規定
- ラベリングおよびパッケージに関する要求事項 医療機器のラベリングとパッケージに関して、ISO 13485:2016 よりもさらに詳細で危険な要求事項を設定している。するための具体的な手順が含まれる。
- その他の追加要望事項
- UDI(機器固有識別)に関する詳細な要求
- トレーサビリティに関する特定の要求事項
- 有害事象報告に関する特定の規定
- 製品回収に関する詳細な要求事項

4.7 要点6.QMSRでは「リスクマネジメント活動」の範囲がQSRよりも広がる
QMSRにおける最も重要な変更点の一つは、リスクマネジメント活動の範囲の大幅な拡大である。 従来のQS規則では、リスクマネジメントは主に設計バリデーションにおいてリスク分析に限定されていた。
ISO 13485:2016の採用により、リスクマネジメントは製品のライフサイクル全体を通じた継続的かつ含むようなプロセスとして逐次評価されるようになった。
- 製品の設計段階からリスクを特定し、分析する
- 製造プロセス全体の乳幼児リスクを継続的に評価する
- 製品の市販後も含めたライフサイクル全体でリスクを監視し、管理する
- リスク分析を静的な一度限りのプロセスではなく、動的で継続的なプロセスとして扱う
この変更は、医療機器の安全性と有効性を総合的に評価するアプローチとして非常に重要な進歩である。製造業者は、製品の概念設計から最終的な廃棄に至るまでの全プロセスにおいて、リスクを継続的に特定、分析、評価、管理、および監視することが求められるようになった。
4.8 QSRとISO 13485におけるリスクマネジメント
QSRにおけるリスクマネジメントの規定は、§820.30「設計管理」の(g)項「設計バリデーション」に関して以下のように定められている:
各製造業者は、手順を確立維持し、設計バリデーションをすることが求められる。設計バリデーションは、定義された運用手順の下で、初期製造のユニット、ロット、またはバッチまたはそれと同様な対象に対して設計バリデーションは、機器が定義された使用者のニーズおよび意図された使用に適合することを保証し、実際のまたは想定した使用条件下での製造ユニットの試験を含むものとする。 設計バリデーションは、適切な場合はソフトウェアのバリデーションおよびリスク分析を含むものとする。 設計バリデーションの結果、例えば設計方法、日付およびバリデーションをした者(一人または複数)を特定するものを設計履歴ファイル(DHF)に文書化することが要求される。
これに対して、ISO 13485:2016では、リスクマネジメントについて以下のように規定している:
7章「製品実現」の7.1項「製品実現の計画」において、組織は、製品実現のために必要なプロセスを計画して、構築することが求められる。その他のプロセスの要求事項と整合性をとる必要があります。
組織は、製品実現におけるリスク管理の一つ以上のプロセスを文書化することが要求される。リスク管理活動による記録は、維持しなければならない(4.2.5参照)。
このように、QSRではリスク管理活動が信頼されているのは設計バリデーションの部分のみであり、リスク分析の実施を要求するのみである。あるいは、7.1項をはじめとする様々な箇条でリスクマネジメント活動を要求している。
ISO 13485の採用により、QMSRではQSRよりも求められるリスク管理の範囲が広がることになります。これは単純範囲の拡大ではなく、リスク管理を製品のライフサイクル全体を通じた継続的なプロセスとして認識へ製品実現の各段階におけるリスクの特定、評価、管理、モニタリングが求められ、これらの活動の記録を維持することが要求される。
このようなリスクマネジメントの要求は、医療機器の安全性と有効性をより確実に保障することを目的としている。で、リスク管理プロセスの透明性と追跡可能性を確保することが意図されている。
4.9 記録に関する規定:要点7.記録
以下の記録の構成要素については、ISO 13485の条項に従うことで達成を得るため、QMSRでは独立した規定を設けていない:
・現設計履歴ファイル(DHF)(820.30(j))については、設計開発の全過程における記録として、ISO 13485の設計開発に関する要求事項の中でカバーされています。
・機器原簿(DMR)(現820.181)については、製品仕様書や製造方法に関する記録として、ISO 13485の製品実現の記録要件の中に含まれています。
・機器履歴簿(DHR)(現820.184)については、個別の製造記録として、ISO 13485の製造及びサービス提供の記録要件でカバーされています。
・品質システム記録(QSR)(現820.186)については、品質マネジメントシステム全般の記録として、ISO 13485の文書化に関する要求事項の中で扱われています。
これらの記録は、ISO 13485の要求事項の中で十分にカバーされているとFDAは判断している。この記録は、管理の重複を避け、より効率的な品質システムの記録を可能にすることを意図したものである。