医療機器の設計・開発・申請における規制要件入門 ~品質、有効性及び安全性の確保~ 6講 医療機器申請と当局査察
承認を早期に取得するためには「申請戦略」から各設計文書を整備する必要がある
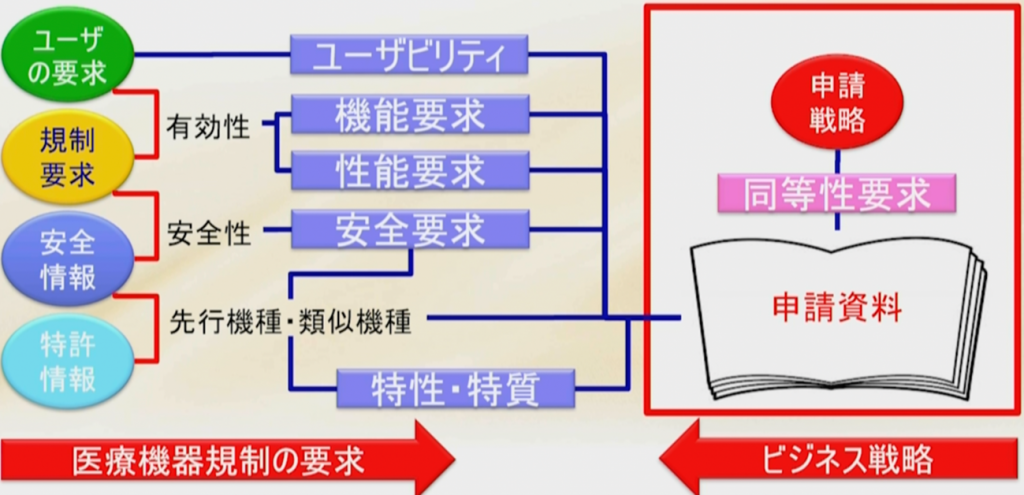
医療機器メーカーには、品目の承認または認証を早期に取得したいという要求があり、どうやれば申請がうまくいくか?早く通せるか?ということに注意が向くものだが、まずやらなければならないことは申請戦略を作ることである。
その申請戦略に基づいて、エビデンスを揃えて、申請資料を作らなければならないのである。
先の章で紹介したように、ユーザーの要求を集め、規制要求を集めて有効性を証明する、安全情報から来る安全要求で安全性を証明する。
さらに先行機種や類似機種に比べて特性・特質があるので、既に世の中に出ている機器または他社品、類似品に対してどんな特徴があるか特性があるかを出す。
新たな特性・特質により、前のものと性能の違う機能を持たせると新たにハザードが生まれ、新たに生まれるリスクをマネージメントする必要が出てくる。
少なくとも先行品に対して同等性を評価しなければならないし、同等以上のものでなければならない。
申請をする段になってこの試験が足りない、あの試験が足りないということにならないように、例えば改良医療機器として申請しようとしているのに新しい機能を付け加えてしまった、それによって新規医療機器になってしまったとか、クラスIIで抑えようと思ったのに、気がついたらクラスⅢになってしまっていた、などということがないようにしなければならない。
方針 (Step1) 〜申請資料〜
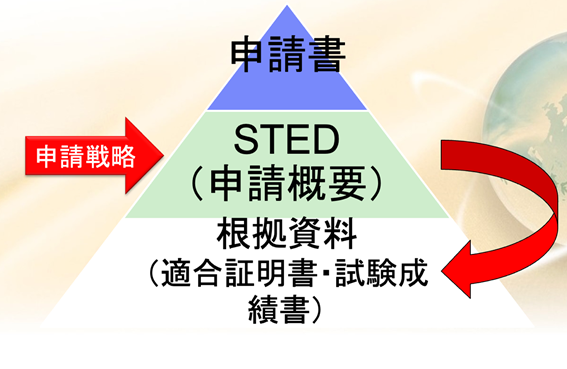
申請資料は、まず鏡として申請書がある。その下にステッドっていうものがある、これが申請概要である。
認証を受ける場合も承認を受ける場合も、このステッドを必ず書かなければならない。
このステッドというのは、サマリー文書のことをいう。
テクニカルドキュメントのサマリー文書は、日本語で言うと申請概要と言われるものである。
設計部門は、色々な設計資料、設計文書や仕様書であるとかまたはテストの記録、または外部試験機関による試験結果、成績証明書等を集めているが、それらすべてを認証申請の時に出すわけではない。
大事なことは、それらを集めた申請概要を書かなければならないということである
その申請概要の書き方、ステッドの書き方というのは当局から出ている。書き方の例というのがでているので、それを参照し作成する。
その根拠資料たるテクニカル文書(設計文書)は、いつ見られるかというと、申請をして、ステッドを見て。当局の方で信頼性調査を選ぶ。例えば二つ選ばれ、この資料を見せてください、次はこの資料を見せてくださいという風にランダムに選ばれる。どの資料を見せてくれと言われるか分からないので、すべての資料を取り揃えておかなければならない。
もし当局からの調査依頼に対して資料がなかった場合、慌てて作ったとしても間に合わない。医療機器の承認審査の期間というのは期限が決められており、その間に必要な資料が出せないと、申請が却下になってしまうということもあるので気をつけなければならない。
こういった根拠資料、多くの仕様書、そしてテスト結果、検証の記録、試験の成績書、これらの物を集めて、それをサマリーした文書がステッドと呼ばれるもので、このステッドを申請戦略に基づいて書くということが申請資料を作るということに相当する。
PMDA申請における調査対象
でここで気をつけていただきたいことなのですけど
ではどういう調査を受けるかということなのですね
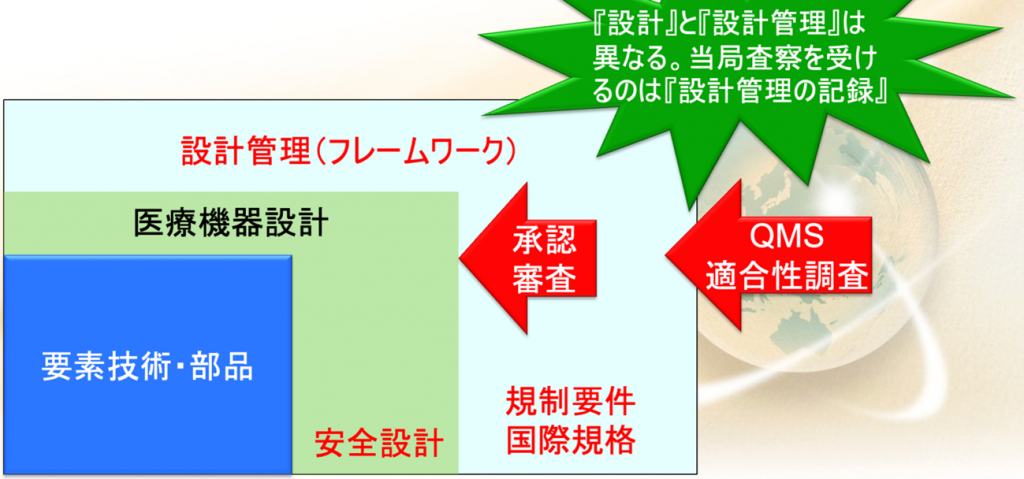
昨今は大学が要素技術を持っていることがある。
例えばソフトウェアのアルゴリズムだけあるなどするのだが、アルゴリズムだけあっても、実際には文書がないので、それでは設計したということにならない。
またもう一つは、この要素技術はどちらかといえば有効性の方で、安全設計はされてないことが多い。
そこで必要なのは図の青色に書いた要素技術をさらに拡大した、医療機器設計、医療機器に大事なことは安全設計なので、グリーンのエリアまで広めなければならないということなのだ。
さらに規制要件とか国際規格に沿ってその設計を管理しなければならない。
これを設計管理(フレームワーク)と言う
後述するが、設計と設計管理の違いを分かってない方が非常に多い。
設計と設計管理は異なるのである。
まず承認や認証で見られるのが、この医療機器設計の部分になる。
特に設計資料の中で、どんな機能で、どういう仕様で、どんな性能で、その結果がどうであって、ということと
もう一つは、安全設計が正しくされて、リスクが回避されている、リスクが低減されているかということである。
これを審査して、当該の医療機器は有効性・安全性がある、という結論になる。
で、もう一つ大事なことは、このQMS適合性調査というのが行われます
これは設計管理が対象になる。
設計と設計管理の違いについて述べる
医療機器の「設計」というのは、その医療機器そのものの有効性・安全性の審査のために必要となる。
一方で「設計管理」というのは、設計計画書を作ったか、計画書を遵守したか、結果が管理され、結果が記録として残っているか、当然内容が改ざんされていないこと、正しいこと
そういうことも管理しなければならない。
設計検証とかデザインレビューも設計審査が行われていて設計審査をする人が力量のない人では意味がない。素人がレビューしても決して品質の良い医療機器にはならないし、安全性の高い医療機器にならない。この設計審査に加わった人は誰であって、力量がどれくらいあるということを管理し記録に残っているかどうか、設計バリデーションを行って、臨床評価を行って、場合によっては臨床試験を行って、正しく管理されているかどうか、記録が残っているかどうか監査する。これをQMS適合性調査という。
「設計」は審査承認されるもの。「設計管理」はQMS適合性調査で査察されるもの、監査されるものという風にご理解いただきたい。
QMS省令で規制されているのは設計管理であって設計ではないので、そこをよく聞き分けていただきたい。
多くの場合は設計管理がうまくいってない。ISO-13485とかQSRとか、またはQMS省令に従った設計管理がうまくできていないということが多いので気をつけていただきたい。
日本の医療機器のクラス分類と承認要件・QMS調査要件
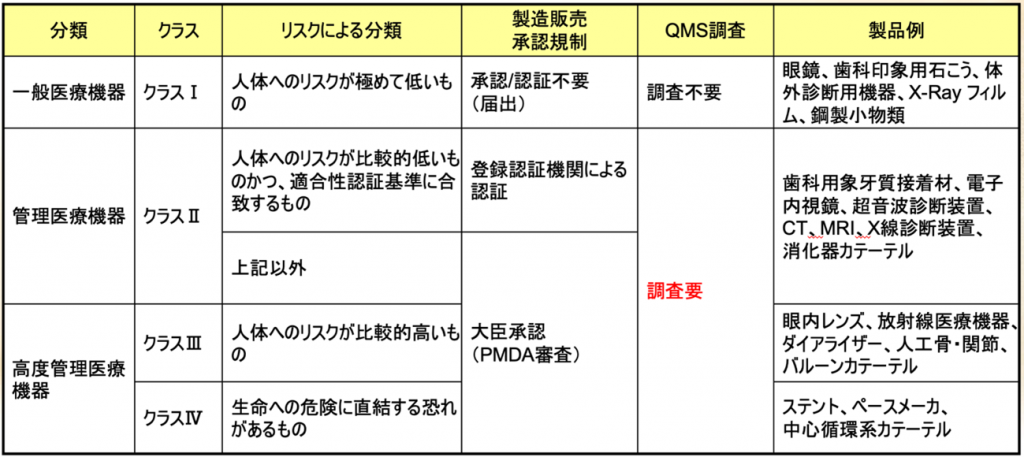
日本の医療機器のクラス分類と承認要件、QMS調査の要件を図でご覧いただきますと
一言です、クラスIを除けば、クラスIIとクラスⅢ、クラスⅣともにQMS適合性調査が必須であるということである。
2015年の薬事法の一部改正によって薬機法になったが、薬機法になって以降、QMS適合性調査は製販に入ることになった。
製販が製造業を監視監督していることを確認する。製販は各製造業者が持っている製品標準書集めて管理しておくことが原則である。
例えばメカは神奈川県で作っている、エレキは群馬県で作っている、ソフトウェアーは千葉県で作っているとする、そうするとメカだけ、エレキだけ、ソフトだけを監査をしても安全性・有効性が確認できない、品質が確認できない。医療機器として組上がって初めて品質が担保されているという事が確認できるので、QMSの適合性調査は製販に入るということになった。
製販に入って、そこから製造業者、これは主に最終組立工場(医療機器としてもう出荷すれば動くものに仕上げる工程のこと)、滅菌前でも相当するし、梱包とか滅菌前のもの、最終医療機器として動作する形にするところに対して実地調査が入ることがある。
実地調査があるかないかというのは当局のみぞ知るということなのだが、ISO-13485を取得していると実地調査が省略されることがある。もちろん省略されない場合も当然ある。
医療機器の民間の第三者機関による認証制度
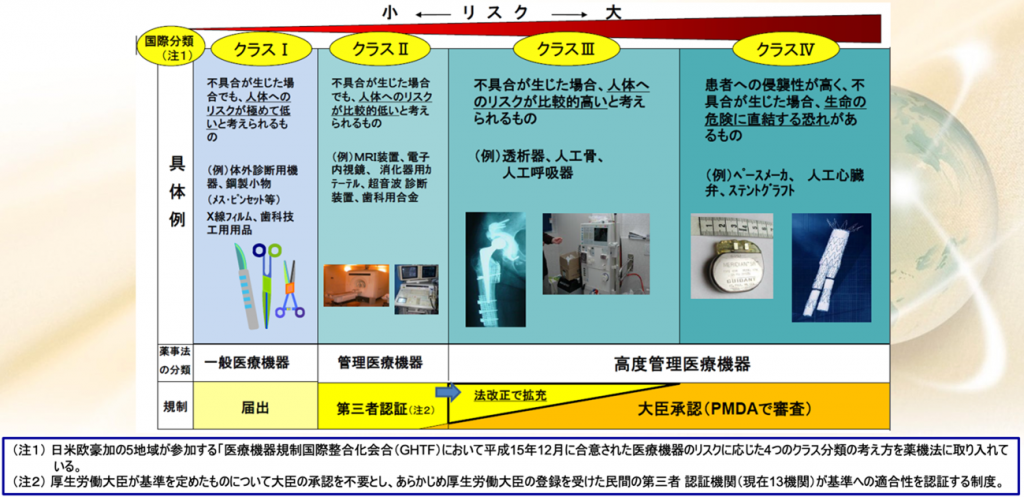
薬機法になってから、医療機器も承認権者が多少変わった。
品目に関しては、都道府県庁の監査というのはなくなった。
管理医療機器クラスIIは主に第三者認証になる。
クラスⅢは大臣承認ですから主にPMDAが審査することになっているが、一部のクラスIIは承認が必要になることがある。
逆に一部のクラスⅢの製品、例えば、もう枯れた製品、コンタクトレンズとかインプラントこういうものはクラスⅢ、要するに侵襲度が高かったり、接触時間が長かったりするのでクラスは高いけれども、安全性が十分に確認されているので第三者認証になったものもある。
一方で認証基準がない新規医療機器に関しては、クラスIIであってもPMDAの大臣承認
になるということもあり得る。日本はこのように承認権者が二つある。国・PMDAと第三者認証機関である。
アメリカは、ほぼ100%を当局FDAが審査をする。
一方でヨーロッパ100%第三者認証機関が認証する。しかもヨーロッパの場合は自己認証が基本で、当局は一切関与しない。
この違いは何かと言うと、承認権者が刑事訴追を受けるか受けないかなのである。
日本とアメリカは承認という制度があるのだが、その代わり、もしそれで万が一何らかの刑事訴追を患者の方から受けることになった場合は、国が成り代わって被告になるというのがこの承認という制度なのである。
一方ヨーロッパでは規制当局が一切この承認という行為に携わらない。 もし事故が起きた場合は当事者間で解決する。裁判を受けて立つというのがヨーロッパの制度なのだ。全て民間が認証するという制度になっており、日米とヨーロッパ欧州では考え方が全く違うということもご紹介しておきたい。
お役立ち動画
医療機器の設計・開発・申請における規制要件入門
~品質、有効性及び安全性の確保~
▶ 1講 医療機器と規制要件
▶ 2講 医療機器の種類
▶ 3講 医療機器と品質
▶ 4講 医療機器と安全性
▶ 5講 医療機器と有効性
▶ 6講 医療機器申請と当局査察










