臨床試験におけるpart11対応
ウェブセミナー 厚労省ER/ES指針関連
臨床試験におけるpart11対応について研究するページです。
*万が一文中に解釈の間違い等がありましても、当社では責任をとりかねます。
本文書の改訂は予告なく行われることがあります。
1.はじめに
近年のITの進化と共に、医薬品の研究開発および製造のプロセスの中でコンピュータシステムが利用される機会が多くなってきた。会社の中を見渡せば、「電子署名」はさておいたとしても、「電子記録」は多くの現場で使用されている。「電子署名」「電子記録」はIT時代のメガトレンドと言えるかもしれない。 しかしながら、製薬企業内の電子システムで、Part 11を遵守したといえるものは皆無であるといっても過言ではないであろう。
FDAによると、「Part11はこの20年間で最も重要なFDAの規則である。」としている。20年間というのは、80年代のオーディット・トレイルの発行にはじまり、Validationに関する規則などを含んだ期間のことである。それらにもましてPart11は重要な規則であると位置づけている。
また、「業界が規則の要求事項に準拠することで技術的に前進していくことを可能にしている。Part11は情報化時代においてデータの完全性を維持するためのCommon Senseアプローチである。」ともしている。
確かに昨今の技術の進歩は目を見張るものがあり、業務の効率化、高品質化において、電子システムの有効活用はもはや常識である。しかしながら、Part11の序文を読むにつれ、FDAは電子システムを相当に疑っているように思えてくる。全体を通して要求していることは、「不正を防止し不正を発見すること」に終始しているように思える。結果的にPart11は、電子システムに対する遵守事項を増加させたことになる。
Part11は発効されて既に5年以上が過ぎているが、欧米の製薬企業の経営者は最近になってようやく、事の重大性を認識するようになってきた。
さらに昨年ICHでStep 4となったeCTD(電子申請)への対応のためには、電子記録を利用しなければならないことは周知の事項である。
本稿では、Part11におけるFDA対応の注意すべき点、考慮すべき点を解説する。
2. Part 11の背景
2.1 Part 11の生い立ちと関連する米国の法律
1990年代初頭に電子署名いわゆるペーパレスシステムへの認識が始まった。これにより、米国製薬工業協会がFDAに電子署名使用についての指針を示すよう申入れを行った。このことがPart 11発行へのきっかけとなったとされる。
一方で、2000年12月末、行政管理予算局(OMB)から発令された政府機関のペーパーワークを除去するための法律「U.S. Government Paperwork Elimination Act」(GPEA:政府書類作業排除法)が出されている。これは、2003年までにサービスや書式をオンラインで利用可能にして書類の提出も電子的に行えるようにすると規定されている。
その他、1998年には「Office of Management and Budget」(政府事務合理化法)が出されており、政府機関にこの法律に従うよう勧告している。
ちなみにFDAに限らず、米国国防総省、財務省、環境保護庁では、「機密性の高い電子業務の実現には、安全性が保証される必要がある」との認識を持っている。
ちなみにPart11の草案段階において、当初は電子署名に関心を持っていたが、議論を重ねるうちにむしろ電子記録が大切であることが分かってきた。
2.2 電子化における課題
Part11が対象とする「電子記録」「電子署名」使用によるユーザのメリットは言うまでもない。
これに対して、利点が逆に問題になる。
- 作成されたプログラムの内容は、容易には確認できない
- 紙の記録、手書きの署名と比べて、「電子記録」「電子署名」は、改ざん(証拠を残さない変更)が容易かつ改ざんの発見が困難になる。
例えば紙の記録を変更する際は、通常オリジナルのデータが読めるように二重線で打ち消し、新しいデータを書き添え、また同時に署名も行う。一方ネットワークに接続されたコンピュータ上の電子データは、誰にも気づかれないうちに、遠隔地から証拠を残さず変更をすることが可能な場合がある。FDAがセキュリティ(アクセスコントロール)に関して執拗に要求事項を記述している所以である。 - 作成時に意図しなかった作業を追加した場合、不測の問題が生じることがある
- システムが複雑になるほど管理、保守の面も含めてコンピュータの信頼性、安全性に多くの問題を含むことになる
3. 製薬企業におけるPart 11対応
3.1 Part 11遵守が必要な製薬企業
Part11は米国の規則であるが、どのような企業がこれを遵守しなければならないかを考えた場合、下記の5つのパターンがあげられる。
この中で、4.では本社のポリシーや手順書を日本法人へおろして来なければならない。しかしながら、これらの手順書を日本でそのまま採用しようとした場合、スキル、経験の面や組織的な問題(例えばコンピュータシステムの監査部門がないか、または人数が限られる)が多く見受けられる。日本でPart11対応を実施する際の、リスクおよびベネフィットプランの作成が必要と思われる。
また5.に関しては、CROの顧客は製薬企業であるが、各企業ごとにPart11対応ポリシーや手順(ドキュメント、様式など)が異なるため、対応の為に困難が想像される。
3.2 Part 11の対応範囲
Part11の要求事項は、企業内情報インフラ、インターネットおよび市販のソフトウェアにまで及ぶ。このような広範囲な対応を行うために、企業側は莫大な費用と、長期間に渡る対応の必要性を懸念している。また遵守要件が具体化していないため、対応方針、新システムへの移行に不安があることも事実である。
さらに対応上の大きな課題として、Part11はすべてのLegacy System(現在使用中でPart 11発効以前に開発されたシステム)についても対応を要求していることがあげられる。FDAはLegacy Systemを免責しない理由として、下記の5つをあげている。
- FDAが電子データシステムへアクセスして、査察を行うことを拒否される可能性がある
- システムに対して許可されないアクセスを許してしまう
- IDやパスワードの再利用を許してしまう
- システムがバリデーションされない状態になることを許してしまう
- 電子記録が多くの方法で改ざんされ、また発見できない状態を許してしまう
1997年8月20日時点にバリデートされた状態にあると判断されたシステムも、バリデートされた状態であり続けるためにはPart11のシステム化要件を組み込むことが必要である。何故ならば、Part 11にはソフトウェアに設計しなければならない特別な要求事項(セキュリティ、オーディット・トレイル等)があるためである。例えば、従来はセキュリティに関してはほとんどの場合、バリデーションの対象とされてこなかったのである。
Legacy SystemにおいてPart11対応を行うためには、下記の事項を考慮する必要がある。
- トレーサビリティーを具備した、ドキュメンテーションの整備
コンピュータシステムの開発、変更管理のためには、各種ドキュメント間の整合性を保つことが求められる。
その目的からTraceability Matrixと呼ばれる管理ドキュメントを作成し、更新する必要がある。
Traceability Matrixに関しては後述する。 - 範囲を狭めるためのサブシステム化
昨今のネットワーク技術を利用して、あらゆるシステム間で電子データの交換が容易に行われるようになってきた。しかしながら規制要件適応のシステム(つまりオーディット・トレイルデータを扱うシステム)が、他の規制要件適応外システム(例えば人事管理システム)等とネットワーク接続して利用されている場合は、本来必要ないシステム(この例の場合は、人事管理システム)までPart 11対応を要求されてしまうと考えられる。したがって、規制要件適応のシステムは出来る限り単体運用とし、ユーザ登録(ID、パスワードの発行)等は当該システム内で閉じて行うようにしなければならない。また出来る限り、社内・外のネットワークからも切り離された状態が望ましい。もちろんこのことは、最新の技術を導入し、業務を効率化するという意図からは大きく後退していることは承知している。 - システムの評価
現在社内で利用しているLegacy Systemを調査し、それぞれにおいてどの程度の追加作業が必要となるかを見積もらなければならない。 - 回顧的バリデーション
Legacy Systemの回顧的バリデーションは、要求および仕様を決めることに始まる通常のProspectiveバリデーションと同じライフサイクルを踏む。このプロセスは通常の開発のProspectiveバリデーションよりも高価になる事もありうる(エラー、バグ、新しい特性の必要性等を発見することがあるため)。
3.3 Compliance Policy Guide (CPG)
FDAは、1999年5月に「Compliance Policy Guide on 21 CFR Part 11」 を発行し、FDAの期待や施行方法を明確に述べている。これはPart11に対するFDAの考え方を示したものであり、査察や是正措置執行に際する手引きとなっている。 CPGのキーコンセプトは “validation of systems to ensure accuracy, reliability, consistent intended performance, and the ability of discern invalid or altered records.”としている。つまり、FDAは「データの一貫性、正確性」に関心があり、また「改ざんを簡単に出来ないようにする」「改ざんされたら分かるようにする」ということを要求しているのである。具体的にはコンピュータ中のデータは、セキュリティで保護されており、なおかつ変更をはじめ操作内容を自動的に記録する機能(Audit Trail)を持たしていないといけないということである。
実際21 CFR Part 11発効以来Security・Adult Trail・Integrityに関する指摘が増加している。
ただしCPGでは、FDAの査察ではオーディット・トレイル対応の観点で判断し、Part11のみでの査察・判断はしないとしている。
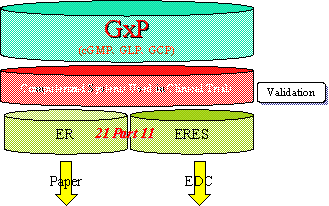
図1 オーディット・トレイルとValidation,ERESの関係
3.4 3つの指針
Part11に対して、下記の3つのステップで対応するべきであると考えられる。
- 運用・手順はすぐに対応する
システムのPart11対応(すなわち再設計、再バリデーション)はすぐには実施が困難がある。しかしながら、Part 11の要求事項の中には、運用により対応できる事項も多い。例えば対応するSOPを作成することによって、下記の事項を規定し実施することが求められる。
① ユーザ・アカウントの管理方法
・ アカウント(ユーザーID等)の再利用、再割り当ては禁止する
・ユーザーIDとパスワードの管理
② パスワードの設定方法
・ パスワード規則を制定する(最小文字数、記号を含めるなど)
・ 定期的にパスワードを変更する
③ ユーザの教育
④ 品質保証体制の確立
・ ID、パスワードの不正使用をレポートする
・ ID、パスワードを作成する装置の初期、定期テストを行う - 新システムはすべて対応する
今後導入するシステムは、Part11に準拠しない限り、使用を開始してはならない。 - Legacy Systemはすべて対応完了する
前述のとおりである。
4 FDAが要求するデータの品質
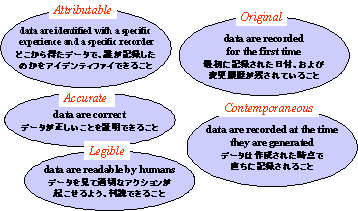
FDAの関心事は、新薬の申請に関するデータの品質である。品質はプロセスに依存しており、プロセスに織り込まれている。申請書中のデータは、電子的に記録されたものであろうと紙に記録されたものであろうと、図2に示すような本質的な品質要素を満足していなければならない。
つまりデータはトレーサビリティーがあり、オリジナルなもので、正確かつ最新で読み易いものでなければならない。トレーサビリティーのあるデータとは、データの観察、記録に責任ある人に辿りつくことのできるデータのことである。自動化システムでは、このトレーサビリティーはすべての入力に責任ある人を特定できるよう設計されたコンピュータシステムで得ることができる。
このことはもはや従来の紙による申請は、事実上困難であることを示唆している。
4.1 Attributable
データはどこで発生し、誰によって記録されたのかを明らかにする。
例) 臨床検査値の血圧は、どの患者のどの訪問回のもので、どのドクターによって記録されたものか。
つまりアプリケーションは、入力した責任者を特定できるように設計されていなければならない。
4.2 Original
データがはじめて記録されたものであり、最初に記録された日付、および変更履歴が残されていなければならない。
4.3 Accurate
データが正しいものであり、最新であること。
4.4 Contemporaneous
観察された時点で直ちに記録されたものであること。
4.5 Legible
人が読むことができるものであること。
5. Guidance for Industry Computerized System Used in Clinical Trials
FDAは1999年4月に「Guidance for Industry Computerized System Used in Clinical Trials」 (以下、ガイダンスと略す)を発行した。これには臨床試験におけるコンピュータ利用の詳細な要求事項が記載されている。
このガイダンスの序文には、「本書は臨床試験の記録に関する積年のレギュレーションを反映するものである。電子記録、電子署名(21 CFR Part 11)の要求事項も反映している。」とある。これは非常に画期的なことであり、ともすると具体性に欠け、対応に混乱をきたしているPart11への対応について理解を深めることができることで価値があるといえる。なおGLP、GMPの各分野においては、Part11以降に出された同様なガイドラインは存在しない。
このガイダンスの序文には、”Persons using the data from computerized systems should have confidence that the data are no less reliable than data in paper form.”(コンピュータシステムによってデータを使用している人は、そのデータが信頼性の点で紙のデータに匹敵することを確信すべきである。)とある。
5.1 FDAの基本原則
FDAが重視するのは、業務プロセスおよびその結果である。「プロセスを無視した結果の達成はありえない」のである。プロセスには電子データシステムの開発および運用段階の作業管理を含んでいる。
つまりFDAの関心ごとは、業務プロセスがコンピュータ化された際に、その品質および 品質保証が劣化してはならないということである。製薬企業は、コンピュータシステムの完全性、正確性、信頼性および一貫したパフォーマンスに対する要求事項に適合していることを保証し、文書化しておく必要がある。
ガイダンスでは基本原則を、下記のように列記している。
A. Identify protocol e-steps
各臨床試験のプロトコルに、どのステップでコンピュータ化されたシステムを使用し、データを作成、改訂、維持、保存、取出し、または送信するかを特定する。
B. Document HW/SW
各臨床試験において、どのソフトウェアとハードウェアが使用するのかを文書化する。この文書は、治験の記録として保存されなければならない。
C. Retain data sources
ソースドキュメントは保持され、臨床試験を再構成でき、評価を可能にする。
FDAは、申請されたデータをもとに臨床試験を再構成できることを期待している。これはデータだけではなく、データの入手方法や管理方法にも適用される。したがって、データや記録の加工に使用される全てのコンピュータシステム、ソフトウェア開発ツール、アプリケーション等は、関係するデータや記録の保存期間中は利用できなければならない。製薬企業はこれらを自ら、またはベンダーと契約して保存する必要がある。
ただしFDAでは「製薬企業やベンダーが旧バージョンのソフトウェアを稼動させる能力を保持することを期待しているが、古いコンピュータシステムを稼動させることが困難な場合があることを認識している。」としている。
またPart11では、査察対応のためだけに旧システムの保持を行わないでいいように、新システムへデータの「正確なコピー」を作成すること(つまりデータの移行)を求めている。ここで大切なことは、データは生データのみならず、メタデータも含むということである。メタデータとは、Audit Trail等で記録された操作履歴、変更履歴等のことである。
D. Direct entry e-source
オリジナルの観察結果が直接コンピュータシステムに入力される場合は、電子記録がソースドキュメントになる。
これはFDAによる”電子生データ”の定義であると理解できる。
E. Parity with paper
コンピュータ化されたシステムの設計は、紙ベースでのシステムと同等に、全ての記録が規制要件を満たしていることを保障するものであること。
臨床試験で使用するコンピュータシステムの設計は、紙ベースの作業(マニュアルの作業)と同程度の信頼性をもっていることを保証しなければならないということである。
F. Study site originals
治験担当医は、治験依頼者またはCROに送付した質門回答文を含む全てのソースドキュメントのオリジナルか、証明付の写しを保持しなければならない。
G. Data change control
何らかの理由により、記録を変更した場合、監査証跡が残され、変更前の情報が読める形で残されていなければならない。
臨床試験の電子データに対して、必要な変更を行った際に、元の情報が曖昧になってはならないのである。変更が実施されたことを明確に記録すると共に、元の情報を検索、閲覧する方法を確立しておかなければならない。
H. e-Audit trail
電子媒体に保存されるデータの変更は21 CFR Part 11に則り、誰が、いつ、なぜ変更を行ったのかを記録した監査証跡が常にとられなければならない。
Part11は電子記録システムの使用者が監査証跡を維持することを求めているが、これは電子記録の信頼性、完全性を保証するものである。監査証跡は、「誰が」、「いつ(作業の日付と時刻)」、「どの電子記録(データ)を何から何に変更したのか」をシステムが自動的に記録するのが一般的である。しかしながら「なぜ変更を行ったのか」を含めなければならないとあるように、理由に関しては操作している人が直接入力する必要がある。
コンピュータシステムでは、監査証跡を確立する以下の2つの方法がある。
1. 記録の1要素が変更されるごとに全体の記録のコピーをとる
2. データ項目レベルでの変更を追跡できる様にする。
もし変更が多い場合は、前者は巨大な記憶容量が必要になり、後者のサイズは小さなものになる。
監査証跡は、臨床データの保存期間と同一期間は保存し、当局の査察時に必要に応じて提出(提示)することができなければならない。さらにFDAは査察官が臨床試験の現場と、製薬企業のデータを保存しているコンピュータ上の両方で監査証跡を要求することがあるとしている。
I. Inspections
FDAは申請に関わるすべての記録を、それらがどのように作成され、維持されているかに関わらず、調査することがある。
FDAは申請に関連するすべての記録を、作成、維持方法に関係なく査察するかもしれないとしている。「維持方法」とは、例えばバックアップされたデータのことである。データのバックアップは通常テープ装置などに吸い上げることがあるが、こういう形式では、FDAが要求する査察時の提示が不可能である。したがって、査察時まではバックアップされた電子記録と監査証跡に関しても、稼動中のコンピュータシステム上に保存しておくことが望まれる。
J. Data Retrieval
データは各被験者に関連付けられた上で、全ての情報が、その被験者まで遡れること。
K. System Design
コンピュータシステムは次のとおり設計されていること。
(1) プロトコルによって要求された全ての事項を、システムが満たしていること。(例えば、データはメートル法で記録される。臨床試験はブラインドで行うなど)
(2) データの作成、改訂、維持、保存、取出し、または送信中に生じたエラーを予め除去する。
L. Security
データやコンピュータシステムに対する不正アクセスを防止するために安全策があること。
セキュリティには、以下のように物理的セキュリティと論理的セキュリティがある。
1. 物理的セキュリティ
ソフトウェアに組込まれたセキュリティに加えて、ハードウェア的な安全対策を実施し、コンピュータシステムおよびデータへのアクセスを許可した者に限定することを保証しなければならない。
例えば、Firewallなどの対策があげられる。
2. 論理的セキュリティ
ユーザがシステムにログオンした後は、データアクセスに制限をかける必要がある。
またコンピュータウイルスに対しても対策が望まれる。
5.2 SOP
ガイダンスでは、コンピュータシステムの使用に関する、例として下記のような6つのSOPが手元にあることとしている。ただしその種類は、これに限定されることはないともしている。(図3参照)
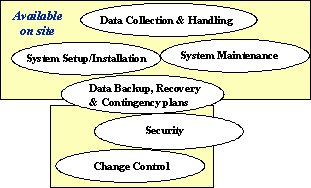
図3 FDA Guidance – 6 Basic System SOPs
5.3 データ入力
5.3.1 電子署名
- 個人がデータ入力を行う許可を所有していることを保証するため、データ入力システムはIDコードとパスワードの組合せまたはバイオメトリック電子署名のような電子署名をデータ入力開始時に必要とするよう設計すること。
- データ入力システムは、また、トレーサビリティーを保証するよう設計すること。すべての変更管理を含む電子記録への入力は入力者の電子署名をもとに行うこと。しかしながら、このことは入力または変更毎に別個の電子署名を入力することを意味しない。例えば電子署名を一度入力すれば複数回の入力や変更が行える。
- 自分自身のパスワードその他のアクセスキーを使用して作業し、これらを他人と共用してはならない。他人のシステムへのアクセスを代行してはならない。
- パスワードその他のアクセスキーは一定期間毎に変更すること。
- ワークステーションを離れる時は、システムをログオフすること。ログオフを怠った場合は、アイドル時間過長で自動的にログオフできることが好ましい。短時間の作業中断に際しては、不正なデータ入力を防止するための何らかの自動プロテクションが必要である。例えば自動スクリーンセイバーでパスワードを入力するまでデータ入力させない方法がある。
5.3.2 オーディットトレイル
- 21 CFR Part 11は電子記録システムの使用者がオーディットトレイルを維持することを求めているが、これは電子記録の信憑性、完全性および、要時秘密性を保護する手順の一つである。
- 確実なコンピュータで作成するタイムスタンプ付きのオーディットトレイルを使用し、電子記録を作成、改訂、削除するためのエントリーと作業の日付と時刻を個別に記録すること。セクションⅡ、定義の「コミット」の項で記載したとおり、耐久性のある媒体に保存した時に記録を作成したとする。
- オーディットトレイルは少なくとも被験者の電子記録(関係する臨床試験データと記録)の保存期間と同一期間は保存し、当局の照査、コピーに供することができなければならない。
- 電子記録を作成、改訂、削除する人がオーディットトレイルを修正できてはならない。
- 臨床試験実施者はオーディットトレイルのオリジナルか、コピーを保存せねばならない。
- FDAの局員は臨床試験の現場と、関連する電子記録が維持されているその他の場所の両方でオーディットトレイルが読めなければならない。
- オーディットトレイルは途切れることなく、時系列的に作成し、新しいオーディットトレイルを既存のオーディットトレイルに上書きできてはならない。
5.3.3 日付/タイムスタンプ
システムの日付と時刻が正しいことを保証する管理方法が手元になければならない。
日付や時刻を変更する能力は許可した人に限定して与える。この人たちには日付や時刻の不一致を連絡しなければならない。日付や時刻の変更は記録しておく。
日付や時刻はローカルなものになるが、年、日、月、日、時、分を含むこと。当局はシステムを第3者に委託して提供された日付や時刻に同期させることを奨励している。
臨床試験に供されるコンピュータシステムは、恐らく異なるタイムゾーンの複数の場所で実施される臨床試験に使用されることになる。この場合、現地のタイムスタンプは遠隔地の異なるタイムゾーンにあるサーバーに計算させるかもしれない。
5.4 システムの特性
- データの直接入力に使用するシステムは品質データの収集を容易にする特性を持つこと。コンピュータシステム内のプロンプト、フラッグ、その他の支援特性を臨床用語の一貫使用と許容範囲を外れたデータについて警報を発するために使用する。迂回したフィールドに自動的にデータを入力する特性は使用すべきでない。
電子的患者日記とe-CRFはユーザーに注釈を記すことを認めるように設計すること。注釈は特別な情報を認めることになり、データ品質を増す。この情報は副作用や予期しない結果が得られた時、役立つことになる。その記録には注釈を加えた人とその日時を明記する。 - データの直接入力に使用するシステムはデータの検査と照査を容易にする特性を含むように設計すること。データタグ(異なる色やフォントの使用、フラッグなど)はオーディットトレイルに記録されている変更や削除がどのデータについて行われたのかを示すのに使用する。
- データの取出し:コンピュータ製品が製造中止になったり、恐らく互換性のない新しいシステムに変更されるかもしれないことを考えると、臨床試験依頼者は旧システムに記録されたデータを取出し、照査する能力を保持しておくことが肝要である。
これは旧システムのサポートを維持することやデータを新システムに転写することにより行える。新システムに移行する時は、臨床試験データとデータの完全性に関する二次的な情報の正確で完全なコピーを作成することが重要である。この情報は例えば、データの取出しに使用するオーディットトレイルとコンピュータの操作方法を含む。報告書を作成するためにデータを操作、検索、抽出する目的で使用するすべてのデータ取出しソフト、手順書、または検索ロジックは記録しておき報告書のために維持しておく。 - 臨床試験の再構成:FDAは臨床試験を再構成できることを期待している。これはデータだけではなく、データの入手方法や管理方法にも適用される。したがって、データや記録の加工に使用される全てのバージョンの、アプリケーションソフト、操作システム、ソフトウェア開発ツールはこれらのバージョンに関係するデータや記録の保存期間中は利用できなければならない。臨床試験依頼者がこれらを自ら、またはベンダーにソストウエアーを稼動させる能力を契約して保存させるかもしれない(必ずしもサポートはしない)。FDAは臨床試験依頼者やベンダーが旧バージョンのソフトウェアを稼動させる能力を保持することを期待しているが、古いコンピュータシステムを稼動させることが困難な場合があることを認識している。
5.5 セキュリティ
- 物理的セキュリティ
システムに組込まれた内部安全策に加えて、外部的な安全策を準備し、コンピュータシステムへとデータへのアクセスを許可した者に限定することを保証することが必要である。
スタッフはシステムの安全対策と許可した者にアクセスを限定することの重要性を十分認識しなければならない。
システムの取り扱いと保存に関するSOPを準備し、不正なアクセスを防止すること。 - 論理的セキュリティ
臨床現場でのデータアクセスは規制をかけログオンされており、安全策とオーディットトレイルを有するシステムのソフトウェアでモニターすること。データは改ざん、ブラウズ、検索や、保護用システムソフトを通らない外部ソフトウェアプリケーションを使用しての報告に使用してはならない。
許可された者の名前、肩書き、アクセス権を常時示す累積的な記録があること。その記録は現場でアクセスできる臨床記録に入っていること。
臨床試験依頼者が臨床試験専用のコンピュータシステムを提供する場合は、そのシステムは当初の目的専用、かつ、バリデートされた目的専用とすること。
臨床試験に使用しているコンピュータシステムが、通常は他の目的で使用しているシステムの一部である場合、臨床試験用ソフトウェアが、非臨床試験ソフトウェアとの予期せぬ相互作用を予め除去するため、論理的、物理的に隔離されていることを保証するための努力を払うべきである。
ソフトウェアのプログラムを一つでも変更した場合、そのシステムを評価し、変更が論理的セキュリティに与える影響を判定しておくこと。
コンピュータウイルスが、臨床試験データとソフトウェアに与える影響を防止、発見、緩和するための管理を実施すること。
5.6 システムの依存性
臨床試験依頼者は、コンピュータシステムが完全性、正確さ、信頼性および一貫したパーフォーマンスに対する要求事項に適合していることを保証し、記録しておくこと。
- システム文書が臨床試験を実施している場所で即時参照可能であること。この種の文書はコンピュータシステムとハードウェア、ソフトウェア、物理環境の関係を全般的に記載していること。
- FDAは、規制対象の会社が保有する、ソフトウェアのバリデーション結果を示す文書を調査するかもしれない。臨床試験依頼者は、要求されればソフトウェアを使用する場所での査察時にこの種の文書を提示する責任がある。臨床試験実施者は普通、ソフトウェアを作成したり修正したりしない限り、バリデーションに責任がない。
- 市販のソフトウェアの場合、バリデーションの大半がそのソフトを書いた会社で実施済みであるべきである。臨床試験依頼者またはCROは、ベンダーによるこの設計レベルのバリデーション文書(バリデーション文書の原文でもベンダーオーディットの文書でもよい)を保有すべきで、自身で機能テスト(テストデータを使用するなどして)も実施しておくこと。既知のソフトウェアの制限事項、問題点、欠陥修正暦を調査しておくこと。
(1)市販で購入でき、(2)一般的な使用目的のために広く使えるよう設計され、(3)無修正で、(4)データの直接入力に使用されていないデータベースやスプレッドシートのソフトウェアという特別な場合、臨床試験依頼者やCROはデザインレベルのバリデーションの記録を持っていなかもしれない。
しかしながら、臨床試験依頼者やCROは自ら機能テストを(テストデータセットなどを使用して)実施し、既知の制限事項や、問題点や、欠陥修正履歴を調査しておくべきである。 - フトウェアバリデーションを説明するのに重要な文書には次のものがある:
ソフトウェアで意図される作業とそれを実施するために意図される方法を記述したデザイン仕様書;デザイン仕様に基づくテスト計画を書いたもので、構造分析と機能分析を含むもの;そして試験結果と、当初のデザイン仕様が満たされていることをその結果がどのように示しているかの評価。
- 市販のソフトウェアの場合、バリデーションの大半がそのソフトを書いた会社で実施済みであるべきである。臨床試験依頼者またはCROは、ベンダーによるこの設計レベルのバリデーション文書(バリデーション文書の原文でもベンダーオーディットの文書でもよい)を保有すべきで、自身で機能テスト(テストデータを使用するなどして)も実施しておくこと。既知のソフトウェアの制限事項、問題点、欠陥修正暦を調査しておくこと。
- 変更管理
ソフトウェアのアップグレード、設備やコンポーネントの入れ替え、新規の装備などのコンピュータシステムに対する変更がデータの完全性やプロトコルの完全性を維持することを保証する、文書化された手順がなければならない。
システムに対する変更によるインパクトの評価と再バリデートの必要性についての意思決定をすること。操作上の限界やデザイン仕様を超える変更については、再ババリデーションが必要である。
すべての変更を記録として残すこと。
5.7 システム管理
- ソフトウェアのバージョン管理
データの作成、収集、維持、送信に仕様するソフトウェアのバージョンがシステム文書に記載のバージョンであることを保証する方法があること。 - 事故対策
コンピュータシステムが壊れた場合に代わりの方法で臨床試験を続けるための事故対策を記載した手順があること。 - 電子記録のバックアップとリカバリー
バックアップとリカバリーの手順の概要をSOPに明記しておくこと。またそれはデータの紛失防止に十分なものであること。
バックアップの記録はSOPで指定された安全な場所に保存すること。保存場所は、典型的な例を挙げると外部か元の記録場所とは異なる建物である。
バックアップとリカバリーのログは保存し、システムの事故に起因するデータの紛失の性質と範囲の評価を容易にすること。
5.8 人の教育訓練
- 資格
データを入力したり加工する人はそれぞれ、割り当てられた機能を発揮するための教育、訓練、経験またはこの適当な組み合わせが必要である。
臨床試験のモニターに責任ある人は、臨床試験を正しくモニターするために必要なコンピュータシステムの使用について、教育、訓練、経験が必要である。 - 訓練
指定された作業を実施するために人を訓練しなければならない。
訓練は資格のある人が必要に応じて継続的に実施するが、その目的はコンピュータシステムや臨床試験実施中のシステムのすべての変更に慣れることを保証することにある。 - 記録
従業員の教育、訓練、経験は記録しておくこと。
5.9 記録の査察
- FDAは申請に関連するすべての記録を、作成、維持方法に関係なく査察するかもしれない。したがって、システムで、正確で完全な記録を査察、照査、コピーに耐えうる、人が読める形と電子的形態の両方の形で作成できること。
- 臨床試験依頼者は、査察対象場所でFDAの局員が電子記録とオーディットトレイルを査察できるハードウェアーとソフトウェアを提供できること。
5.10 電子署名の証明
21 CFR 11.100(c)で要求されているとおり、 FDAの署名の要求に適合する電子署名を使用する人は、使用前あるいは使用時に当局に対して1997年8月20日以降にシステムで使用する電子署名は従来の手書きの署名に相当する法的拘束力を持たせる意図があることを証明しなければならない。
21 CFR 11.100(c)に記載のとおり、証明書は従来の手書きの署名のある紙の形で、the Office of Regional Operations (HFC-100), 5600 Fishers Lane, Rockville Maryland 20857に提出すること。
証明書は電子署名の使用に先だって、または使用する日に提出する。しかしながら、一枚の証明書が特定の組織の人が使用する全ての署名をカバーするかもしれない。この証明書は人が作成する法的な記録で、電子署名が従来の手書きの署名と同じ法的意味を持つことを認めるものである。受け入れ可能な証明書のフォームは次のようなものである。
『連邦法タイトル21セクション11.10に従い、ここに【組織名】は世界中のすべての従業員、代理人、代表がおこなうすべての電子署名が従来の手書きの署名と同等の法的拘束力があることを証明する。』
6 Part 11とComputerized System Validation(CSV)の関係
バリデーションは色々な業界で採用されている明確な要求事項である。Part11は電子記録を保持、あるいは電子署名を使用するシステムにいくつかの特徴的な要件をつけ加えた。
システムはバリデートされるまで使用されるべきではない。もしPart11に準拠していないならばバリデートされていないことになる。(システムは要求事項や仕様を満たしていない)
Part11以降も FDAでは、「Computerized System Validationに対する FDAの期待は変わっていない。バリデーションの定義を満たさなければならない。」としている。
Part11において、Closed Systemの一番目の要件としてバリデーションをあげている。
| 11.10 Controls for Closed System Maintenance of Reliability, Integrity,Confidentiality for Electronic Records (a) Validation of systems to ensure accuracy,reliability, consistent intended performance, and the ability to discern invalid or altered records. (正確性,信頼性,意図した性能の一貫した確保,ならびに無効となったり変更されたりした記録を識別する能力が保証されるようにするためのシステムのバリデーション) |
7 Software Development Life Cycle (SDLC)
多くの製薬企業の情報システム部門には、いわゆる「開発標準」という規約文書が存在している。多くの場合開発標準は、ソフトウェア開発のライフサイクルに基づいて記述されている。しかしながら、CSVの観点で全ライフサイクルが網羅的に記載されているかどうかは疑問が残る。
FDAでは、査察官自体がこのSDLCのメソドロジーに基づいて教育を受けている。今後のPart11査察においては、このSDLCに則ったドキュメントの整備と整合性がチェックされるものと思われる。
一般的に、SDLCおよび各フェーズにおけるドキュメンテーションは、下記のようなものが考えられる。
7.1 プランニング (Validation Plan)
このステップでは、バリデーション計画書を作成する。バリデーションに携わる組織、人員構成、期間、必要な文書類等の定義をはじめ、当該システム導入プロジェクト全体の品質保証証計画(Quality Plan)を明らかにする。またプロジェクトの各段階で発生する変更要求に対する処理基準および手順を記載したChange Control Planも作成が必要である。
7.2 リクワイアメント (Requirements Documentation)
User Requirements Specification(URS)を作成する。URSには、ユーザの要求事項を記載し、実際の業務で必要とする機能や、データ、操作環境を定義づける。このなかには、Functional Risk Assessmentと呼ばれる、要求される各機能が正常に動作しない場合や、導入が実現できない場合の業務に対するインパクトの程度やリスクの記述を含める場合がある。
7.3 ベンダー選定 (Vendor Audit Report)
URSにもとづき、Request for Proposal(RFP)を作成する。RFPは、ソフトウェアベンダーへの要求仕様書である。またベンダーからの提案を受け、そのベンダーを査察(Vendor Audit)する場合がある。Vendor Auditの目的は、そのベンダーが要求に合致した品質でソフトウェアの開発を行うことができるのかどうかを、調査することである。またソフトウェアがGxP基準に精通した専門家によって開発され、テストされ、かつ適切に保存されていることをメーカーに確認する。この際に事前にVendor Audit Check Listを作成する必要がある。また実施後のVendor Audit Reportおよびベンダー選定の報告書が作成される。
7.4 設計 (Design Specifications and Reviews/DQ)
ハードウェア、ソフトウェア、必要に応じてネットワークのハイレベルな仕様を記載したDesign Specificationを作成する。またURSをシステム上で実現するソフトウェアの機能仕様を記述したFunctional Specificationと、ハードウェアおよびネットワークの仕様を記述したTechnical Specificationも作成される。
システムのインストール計画書、インストール手順書(クライアントおよびサーバー)、バックアップ/リカバリー手順書もこの段階までに準備しておく必要がある。
URSに対して、設計の妥当性を評価・検証することをDesign Qualification(DQ)と呼ぶ。またDQ報告書も作成されなければならない。
7.5 構築、システムテスト (Build, Code Reviews, System Testing)
コーディング(プログラミング)を実施する場合は、詳細設計書、プログラム設計書等が作成されるのが一般的である。
パッケージ導入の場合は、カスタマイズまたはコンフィギュレーションを行う。コンフィギュレーションとは、パッケージをカスタマイズするのではなく、パラメータ等によって、そのアプリケーションの設定を変更できる機能である。コンフィギュレーションによって設定の変更が可能なソフトウェアを、コンフィギュアラブル・アプリケーション(Configurable Application)と呼ぶ。
さらにテスト仕様書が作成された後、テストを実施し、テスト結果報告書等が作成される。
7.6 導入 (Installation and Configuration Documentation)
作成された手順書をもとに、ハードウェアの設置、OSやデータベース等のインストレーション、ソフトウェアの導入を行い、その結果をインストレーション結果報告書として作成する。
7.7 クオリファイ (IQ/OQ/PQ)
この段階を説明するには、GAMPのガイドラインによる「V-Model」(図4参照)が有効である。Technical Specificationに対し、準備されたプラットフォームが正確に稼動し、その要求を満たすことを検証するテストをInstallation Qualification(IQ)と呼ぶ。
またSystem Specificationに対して、要求された機能が実現されていることを検証するテストをOperation Qualification(OQ)と呼ぶ。
さらにユーザの要求仕様であるURSに対して、要求された要件が実現され、稼動できることを検証するテストをPerformance Qualification(PQ)と呼ぶ。しばしばこのPQは、その名前から、コンピュータシステムやネットワークのパフォーマンス試験のことと誤解されることがあるが、そうではない。ユーザの業務プロセスが、コンピュータ化により要求に応じて滞ることなく実現できるかを検証するのがPQの目的である。従って、PQはユーザが実施することになる。
これらIQ、OQ、PQに対する計画書(Scripts、テストデータを含む)および実施結果報告書がそれぞれ作成される。
また前の導入段階およびこのクオリファイの段階で発生した不具合は別途Incident Logとして記録され、Change Management Planに従って、変更管理が実施される。さらに変更はChange Logとして記録される。
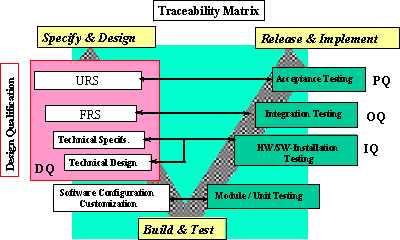
図4 V-Model
7.8 デプロイメント (Deployment)
あらかじめDeployment Planを作成し、それに従ってユーザへの展開を図る。またシステム導入後の新しい業務プロセスを記述したSOPは前もって準備されなければならない。データを入力または加工するユーザは、システムを正確に使用するための教育、訓練を受けなければならない。教育は資格のある人が、必要に応じて継続的に実施する。大切なことは、単にシステムの操作手順ではなく、新SOPに沿った教育が実施されなければならない。また特にシステムが変更された場合には、変更後のシステムに十分慣れる事が重要である。教育に関する文書として、Training Plan、カリキュラム、ユーザの教育記録等がある。
またこの段階で作成される文書として、システムをサービス(運用サポート、メンテナンス、バックアップ等)する側とサービスを受ける側(ユーザ)がその利用形態(サポート体制、稼働時間、ヘルプデスク、ユーザの責任等)について合意したService Level Agreement(SLA)がある。
さらにシステムの不慮の事故に備えて、Disaster Recovery Planを検討する必要がある。
>7.9 廃止 (Decommission)
医薬品の開発期間に比べ、コンピュータシステム(H/WおよびS/W)の寿命はあまりにも短い。一連の薬剤開発期間中にコンピュータ製品が製造中止になったり、互換性のとれない新しいシステムにバージョンアップされることも考慮すべきである。
現行のシステムをリタイアさせ、新システムに移行するためには、正確なデータの移行(Data Migration)を実施する必要がある。その際に、旧システムに蓄えられた監査証跡(操作・変更履歴)を含めて移行が行われなければならない。
データの移行に使用するプログラムは、同様にバリデーションされていなければならない。また移行作業の手順書を作成し、実施を記録したデータ移行結果報告書が作成されなければならない。
7.10 変更管理
上記のライフサイクルの中で、変更管理は非常に重要である。変更に際して、各段階にあげた多くの文書間の整合性を完全に保つことはそうたやすくはない。ガイダンスでは、ユーザによる要求事項の変更など、色々な場面におけるシステムの変更の管理を十分に行うことを要求している。そこで、文書間の整合性をトレースするためのテーブルを常に作成し、メンテナンスすることが要求される。この文書のことをTraceability Matrix(図5参照)と呼ぶ。これはシステムの開発または導入中に作成される各種のドキュメントに関して、1つの変更要求がどのように関係し、管理、維持されるか、またどのように変更がテストされ結果が管理されるかをまとめた文書である。たとえば、ユーザの要求仕様(URS)の何章の何番が、Functional Specificationの何章の何番に対応し記載され、テスト仕様書の何章の何番に対応し、テスト結果報告書の何章の何番に記載されているかを明らかにするのである。
多くの場合、ソフトウェア開発は成り行きに任せ、後半の文書ほど現実を現しているが、前の段階の文書がそれに呼応して変更されることは少ないようである。製薬企業各社は、現在使用しているシステム(いわゆるLegacy System)について、このTraceability Matrixを作成することが望まれる。
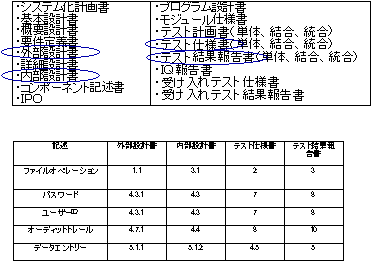
図5 Traceability Matrix
8 市販パッケージにおけるバリデーション
筆者は長年にわたり、臨床試験管理システムや臨床データ管理システム等のアプリケーション・システムを開発、サポートしてた経験を持つ。その際にある製薬企業の情報システム部員から、「パッケージシステムをカスタマイズしないで導入するのだから、バリデーションは不要ではないか」という指摘を受けたことがある。つまり彼は、バリデーションとはコンピュータシステムの導入テストあるいはソフトウェアの不具合(バグ)の発見のことであると考えているのである。これは言うまでもなく間違いであり、その理由を解説してみたい。
FDAは単にコンピュータシステムにバグや不具合のないことを要求しているのではない。紙ベースのオペレーションがコンピュータ化された際に、申請に関係するデータの品質および品質保証が、劣化しないという保証を求めているのである。FDAにとってみると、製薬企業がコンピュータシステムを使用しようがしまいが問題にはしていない。ましてや、何処の何という製品を利用しているかということは関心がないのである。業務をコンピュータ化することによって、何らかの問題が発生しないかという懸念を払拭することが重要であるといえる。
ソフトウェア自体のバリデーションは外部企業やベンダーによって実施されるものが多いが、そのソフトウェアの業務への適合性についての最終的な責任は製薬企業にある。またそのバリデーションの適合性を評価できる記録は、製薬企業で保管する義務がある。FDAは、システムの開発期間中と、リタイアまでの稼動期間中の両方についてそのライフサイクルを管理することを求めている。
8.1 市販パッケージを利用することの利点
前述のとおり、URSにおいて、ユーザはコンピュータシステムで実現したい機能を定義する。またそれらの要求をFRSに具体化する。パッケージを導入し、業務への適合を図る場合、FRS作成時には次の4つの場合分けが考えられる。
- 業務プロセスのうち、パッケージを変更せずにそのままの機能利用する
- パッケージの機能を、パラメータを設定することにより変更する(コンフィグレーション)
- パッケージにない機能を追加製作する、あるいはカスタマイズする
- 業務プロセスに関係しないため、パッケージの機能を使用しない。
これらのうち、FRSを記述しなければならないのは、2.と3.である。大部分をパッケージの機能そのままで利用できる場合には、FRSの記載を最小限にとどめることができ、OQの対象を限定することができる。
良く「バリデーションを軽減するために、パッケージを導入する」といった考え方を耳にするが、実際にはこのような考え方により、バリデーションのドキュメンテーションおよびOQ作業が軽減できるのである。
なお、上記3.はさらに、Design Specificationを作成しなければならないことを申し添えておく。
8.2 バリデーションの優先順位付け
あまりにも品質を追い求めることにより、いつまでたってもシステムが稼動できないケースがある。バリデーションで必要なことは、その機能が業務プロセスに与えるインパクトや、正常に動作しない場合のリスクを調査し、優先順位をつけることである。
あまりにもリスクのない機能まで、バリデーションの対応とすれば、莫大な費用と時間がかかってしまう。
前述した、Functional Risk Assessmentでは、パッケージの各機能とユーザの業務プロセスのマトリックスを作成し、その交差するセルに、High Risk、Middle Risk、Low Riskといった評点を付与していく。
ここでHigh Riskとされたものから優先順位をつけ、バリデーションを実施していけば、効率的である。
Middle RiskおよびLow Riskについては、余裕があれば対応するくらいの気持ちでよいのではないかと考える。
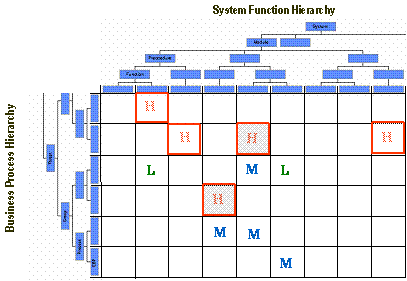
図6 Functional Risk Assessment
9 品質管理と品質保証
FDAが製薬企業に要求することは、ひとえにFDAに提出されるデータおよびドキュメントの品質であり、それらの品質保証である。医薬品の研究開発および製造で扱うデータは、その医薬品の安全性、有効性、品質に大きく関係するものであるからである。つまりそれらは人の健康に幅広く関係するもので、最高の品質と完全性がなければならないのである。
9.1 データの信頼性確保
第3回ICH横浜会議では、臨床データの質向上のためにQC(品質管理)とQA(品質保証)が大きく取り上げられた。概して日本での品質改善活動では、QCが執拗に行われているが、QAに関しては、意識が浅いとされている。
1997年3月13日に発行された「医薬品の臨床試験の基準」(中薬審第40号)および同年3月27日「医薬品の臨床試験の実施の基準(GCP)」(厚生省令第28号)- いわゆるGCPによると、治験依頼者の責務は『品質保証システムの履行と保持』にあるといえる。
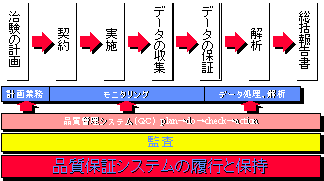
図7 臨床開発における品質保証システム
9.2 CSVにおけるQA部門の役割
SDLCにおいて作成される各文書は、各段階の適切な時点でQA担当者によるレビュが実施されることが必要である。日本においてはCVQA(Computer Validation Quality Assurance)の担当者の育成が遅れている。
各ドキュメントがタイムリーにレビュされ、その品質が保証されないまま、SDLCのステップを移行することはリスクを伴う。なぜならば、たとえばSDLCの最終局面において不備の指摘があった場合には、それまでの労力と時間を無駄にし、再度同様なプロセスを実行しなければならなくなるからである。
CSVに関する手順書およびドキュメントの定義は、あらかじめ用意されているべきであり、QAによるレビュも完了していなければならない。
9.3 品質管理とは
品質管理とは、顧客の要求を満足させる品質をもった製品を作り出し、顧客に提供する機能をいう。日本では良くこのQCのことをQuality Checkと勘違いされているように感じる。これは間違いであり、Quality Controlが正しい。日本の多くの製薬企業においては、データの入力に際して、ダブルエントリーを用い、また読みあわせを数回行うなどして入力のミス等を修正している。しかしながらこの作業はあくまでもCheckであり、QCでいうところのControlではない。CheckとControlの違いは明らかである。Controlとは、SOPなどの標準(書)に沿って業務プロセスが管理運営されており、それらを逸脱していると正常に戻す機能のことを言う。新薬申請後の書面調査においても同様にCheckが行われているが、プロセスに関する品質管理はレビュされていないようである。これは日本と米国での考え方の大きな違いであるといえる。
ちなみに、ManageとControlは邦訳するとどちらも「管理」となるが、その違いはこうである。Manageは野球の監督に例えられ、Controlは投手に例えられる。つまりManageとは、全体を指揮、統制(例えば敬遠を指示)する役割であり、Controlとは支持されたとおりに業務を遂行する(例えばストライクを投げる)ことである。
このことから分かるように、QC担当者の役割は、部場において業務プロセスがSOPから逸脱した(またはしそうな)場合において、それを指摘し、正しいプロセスへと誘導することである。決してデータの入力ミスを見つけてあげることではないのである。一般にデータの入力ミスを入力に責任を持つ部門ではなく、他の部門(この場合はQC部門)が修正しているとすると、プロセス中のデータの品質は向上しにくくなる。原則は、データソース側がデータの品質に責任を持つことである。
9.4 品質保証とは
JIS においては、「消費者の要求する品質が十分に満されることを保証するために、生産者が行なう体系的活動」 (JIS Z 8101) と定めている。
QCでの勘違い同様、日本では良くこのQAのことをQuality Auditと勘違いされているように感じる。日本の多くの製薬企業においては、承認された成果物に対してRetrospectiveに抜き取りによって監査が行われるのが一般的である。この方法では、非監査部門が行ったCheckを繰り返していることになる。海外から、日本ではQA部門がQCを行っていると揶揄されることもある。皮肉にも日本の製薬企業が申請するデータは、Checkを繰り返し行っていることから、最も精度が高いとされている。しかしながら、業務プロセスの品質保証がなされていないと指摘されているのである。
当然のことながら、QAはQuality Assuranceのことである。QAで大切なことは、システムの企画・設計・開発・運用の各フェーズにおいて、第三者の観点から規制要件、自社のポリシー、手順書と照らし合わせてレビュすることである。システムの完成後に監査を行い、不足事項を指摘しても、それでは後の祭りである。
QA部門は、非監査部門と協力し合って、システムのライフサイクルの節目節目で、プロセスの品質保証を与えることが大切である。
10 あとがき
製薬企業が Part11に対応するためには、下記に掲げるような4つの事項を考慮しなければならない。
また自社内の努力のみで何とか対応しようとするのではなく、外部のコンサルタントを含めたスキルミックスで対応することが望まれる。
本文中にも述べたが、 Part11の対応は、ともすると長期間、莫大な費用を費やしかねない。それを防止するためには、全社をあげて戦略的な対応法を検討する必要がある。
またFDAの査察官に対して、毅然と対応できる人材の養成も望まれる。