
医薬におけるバリデーション
イーコンプライアンス ウェブセミナー
医薬におけるバリデーションについて研究するページです。
*万が一文中に解釈の間違い等がありましても、当社では責任をとりかねます。
本文書の改訂は予告なく行われることがあります。
本邦では、医薬品の品質確保について、「医薬品及び医薬部外品の製造管理及び品質管理規則」(平成16年12月24日、厚生労働省令第179号、以下GMP省令)が定められており、この規則の中にバリデーションに関する条項がある。
またバリデーションの施行に関し、「バリデーション基準」(平成17年3月30日、薬食監麻発第0330001号)が通知されている。
さらにICHで作成された「原薬GMPのガイドライン」(平成13年11月2日、薬発第1200号)が公布されている。「原薬GMPのガイドライン」により、「構造設備のバリデーションとは、適格性評価(Qualification)を行うことである」とその定義が明確になった。
コンピュータシステムのバリデーションは、GMP省令や、バリデーション基準等に記載はないが、多くのプロセスがコンピュータ制御であることを認識すべきである。
バリデーションにとって重要なことは、「文書化」である。第三者が当該文書を見て、その品質および品質保証を確認できるものでなくてはならない。これを「対監査性」という。
文書がないということは、保証ができないということである。つまり文書は品質および品質保証の証明となるのである。
「あらかじめ」という言葉も重要である。品質保証のためには、あらかじめ定めた仕様や品質がなければならない。
品質保証の基本は、計画のとおりプロセスを遂行し、あらかじめ定めておいた仕様や品質の結果を、繰り返し出力できなければならないのである。
「たまたまやったら、たまたま良い結果が出た。」では、再現性がなく、品質の保証は出来ないのである。
研究室で製剤し、臨床試験(治験)に用いた薬剤を工業化し、工場で生産するわけであるが、治験時に使用したものと同じ品質の製品が大量に継続して生産できるといった保証をとらなければならないのである。
医薬におけるバリデーションとは
FDAが1987年に発行した「Guidelines on General Principles of Process Validation」には医薬におけるバリデーションの定義として以下の記載がある。これはISO-9000の定義を参考にして医薬品向けに変更したものである。
“Establishing documented evidence which provides a high degree of assurance that a specific process will consistently produce a product meeting its predetermined specifications and quality characteristics.”
(文書化された証拠を確立してゆく作業であり、これはあらかじめ定めた仕様や品質にあった製品を継続的に生産するプロセスに対して、高度の保証を与えるものである。)
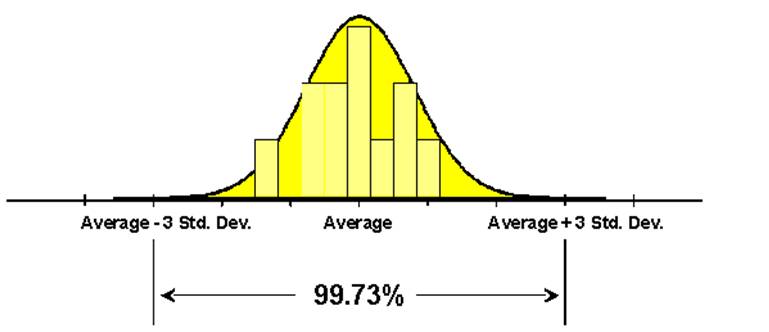
図1.医薬におけるバリデーション
製品品質の分布
以下の図2.では、品質が常時変動している。また平均が上下している。変動も増減している。
全変動(Total Variation)も時間とともに増加している。
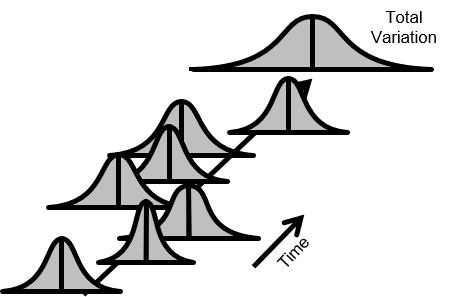
図2.不安定なプロセス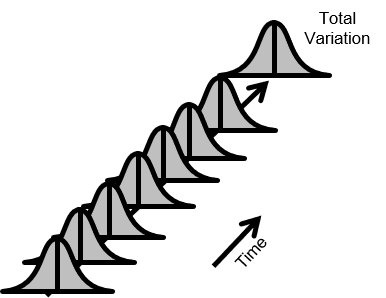
図3.安定なプロセス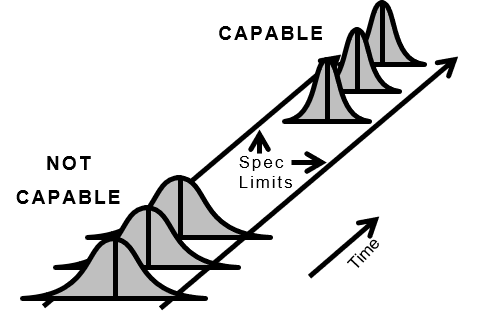
図4.良好な製品を首尾一貫して生産するプロセス
GMPにおけるバリデーションの種類
バリデーションの実施時期
では、バリデーションはいつ実施しなければならないのかというと、GMP省令第13条に以下のような記載がある。
第十三条 製造業者等は、あらかじめ指定した者に、手順書等に基づき、次に掲げる業務を行わせなければならない。
一 次に掲げる場合においてバリデーションを行うこと。
イ 当該製造所において新たに医薬品の製造を開始する場合
ロ 製造手順等に製品の品質に大きな影響を及ぼす変更がある場合
ハ その他製品の製造管理及び品質管理を適切に行うために必要と認められる場合
二 バリデーションの計画及び結果を品質部門に対して文書により報告すること。
2 製造業者等は、前項第一号のバリデーションの結果に基づき、製造管理又は品質管理に関し改善が必要な場合においては、所要の措置を採るとともに、当該措置の記録を作成し、これを保管しなければならない。
まず、製造所を新設した場合や、既存の設備であっても、新たな医薬品の製造を開始する場合にバリデーションが必要である。
また、GMPハードやGMPソフトに対して、品質に影響を与える変更を行った場合も、同様にバリデーションを実施しなければならない。
バリデーションの文書(例:バリデーション計画書、バリデーション報告書など)は、必ず品質部門に提出し、最終的に品質部門が承認しなければならない。
バリデーション基準
平成17年3月30日に「薬事法および採血および供血あつせん業取締法の一部を改正する法律の施行に伴う医薬品、医療機器等の製造管理および品質管理(GMP/QMS)に係る省令および告示の制定および改廃について」(平成17年3月30日、薬食監麻発第0330001号)という課長通知が発出された。
この課長通知によって、「コンピュータ使用医薬品等製造所適正管理ガイドライン」(平成4年2月21日薬監第11号。以下「旧ガイドライン」)が廃止された。また「バリデーション基準」が改定された。
「バリデーション基準」は、本通知の第3章 医薬品・医薬部外品GMP省令の第4に記載がある。
この中で、医薬品・医薬部外品GMP省令に規定するバリデーションについては、「バリデーション基準」および「バリデーション基準の運用について」に基づいて実施することとされた。
バリデーションの目的として、「バリデーションは、製造所の構造設備並びに手順、工程その他の製造管理および品質管理の方法(以下「製造手順等」という。)が期待される結果を与えることを検証し、これを文書とすることによって、目的とする品質に適合する製品を恒常的に製造できるようにすることを目的とする。」との記載がある。
また、実施対象としては、
ア.製造工程
イ.製造を支援するシステム
ウ.洗浄等の作業
があるが、イ.およびウ.については、設備又は機器単位毎に実施しても差し支えなく、また、ウ.については、合理的な根拠に基づき、指標となる成分のみをもって評価しても差し支えないとしている。
GMP省令におけるバリデーションの定義
バリデーションとは一体何であろうか。GMP省令の第2条 定義に、以下の記載がある。
第二条 定義
5 この省令で「バリデーション」とは、製造所の構造設備並びに手順、工程その他の製造管理及び品質管理の方法(以下「製造手順等」という。)が期待される結果を与えることを検証し、これを文書とすることをいう。
GMP省令におけるバリデーションの定義は、「製造所の構造設備」(GMPハード)と「手順、工程その他の製造管理及び品質管理の方法」(GMPソフト)によって、あらかじめ設定した期待の通りの結果を出すことを保証し、文書化することである。
バリデーション基準と適格性評価
我が国では、医薬品の品質確保について、医薬品及び医薬部外品の製造管理及び品質管理規則(平成16年12月24日、厚生労働省令第179号、以下GMP省令という)が定められており、この規則の中にバリデーションに関する条項がある。
バリデーションの施行に関し、バリデーション基準(平成17年3月30日、薬食監麻発第0330001号)が改定された。
また、ICHで作成された「原薬GMPのガイドライン」(平成13年11月2日、薬発第1200号)が発出された。
原薬GMPのガイドラインにより、構造設備のバリデーションとは「適格性評価評価(Qualification)を行うことである」とその定義と内容が明確になった。
製薬分野におけるプロセスエンジニアリングは、プラントなどの設計品質や設備・装置・機械などの性能、試運転から保証運転終了までの製品品質を左右し、その後の運転期間中における保守保安に大きく影響する工学分野である。
適格性評価は、製品品質に直接影響する要因についてのみ、設計段階でDQを、製作・施工段階でIQを、試験・検査・試運転段階でOQとPQを行うことである。
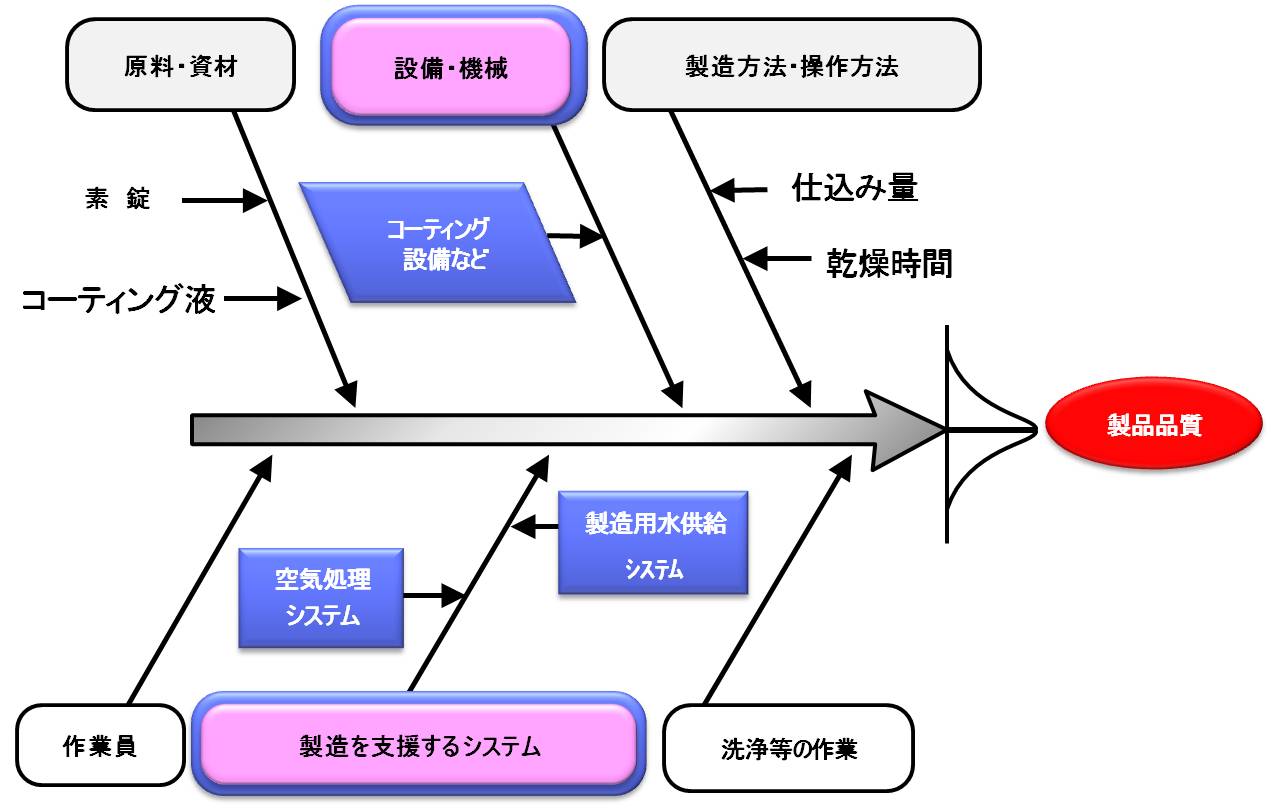
バリデーション基準によると、バリデーションの目的は「製造所の構造設備並びに手順、工程、その他の製造管理および品質管理の方法(以下、製造手順等)が期待される結果を与えることを検証し、これを文書とすることによって、目的とする品質に適合する製品を恒常的に製造できるようにすること。」である。
バリデーションの実施対象は、
- 製造工程
- 製造を支援するシステム
- 洗浄等の作業
2.と3.は、設備または機器単位ごとに実施してもよい。3.は、合理的な根拠に基づき、指標となる成分のみを持って評価してもよい。
「バリデーション」と「適格性評価」との関係
「原薬GMPのガイドライン」によると「原薬の品質及び純度に関して重要であると判断される作業に適用されるバリデーション」のなかの一部として、「プロセスバリデーション(PV)の作業を始める前に、重要な装置及び付帯設備の適格性評価を完了すること。」とある。
すなわち構造設備を対象とするバリデーションを「適格性評価」と呼び、DQ、IQ、OQ、PQから構成される。
「バリデーション」と「適格性評価」との違い
バリデーションはソフトとハードの両方を含み、ハードに関するバリデーションは適格性評価(Qualification)を行うことである。
2011年1月13日に改定版が発表され同6月30日から施行される「EU GMP ANNEX 11 Computerised System」の原則には、「The application should be validated; IT infrastructure should be qualified.(アプリケーションはバリデートされていなければならず、ITインフラストラクチャは適格性が確認されていなければならない。)」との記載がある。
適格性評価は、DQ(設計時適格性評価)、IQ(設備据付時適格性評価)、OQ(運転時適格性評価)、PQ(性能適格性評価)から構成される。
日本のバリデーション基準では、DQについて明確ではない。
適格性評価とプロセスバリデーション
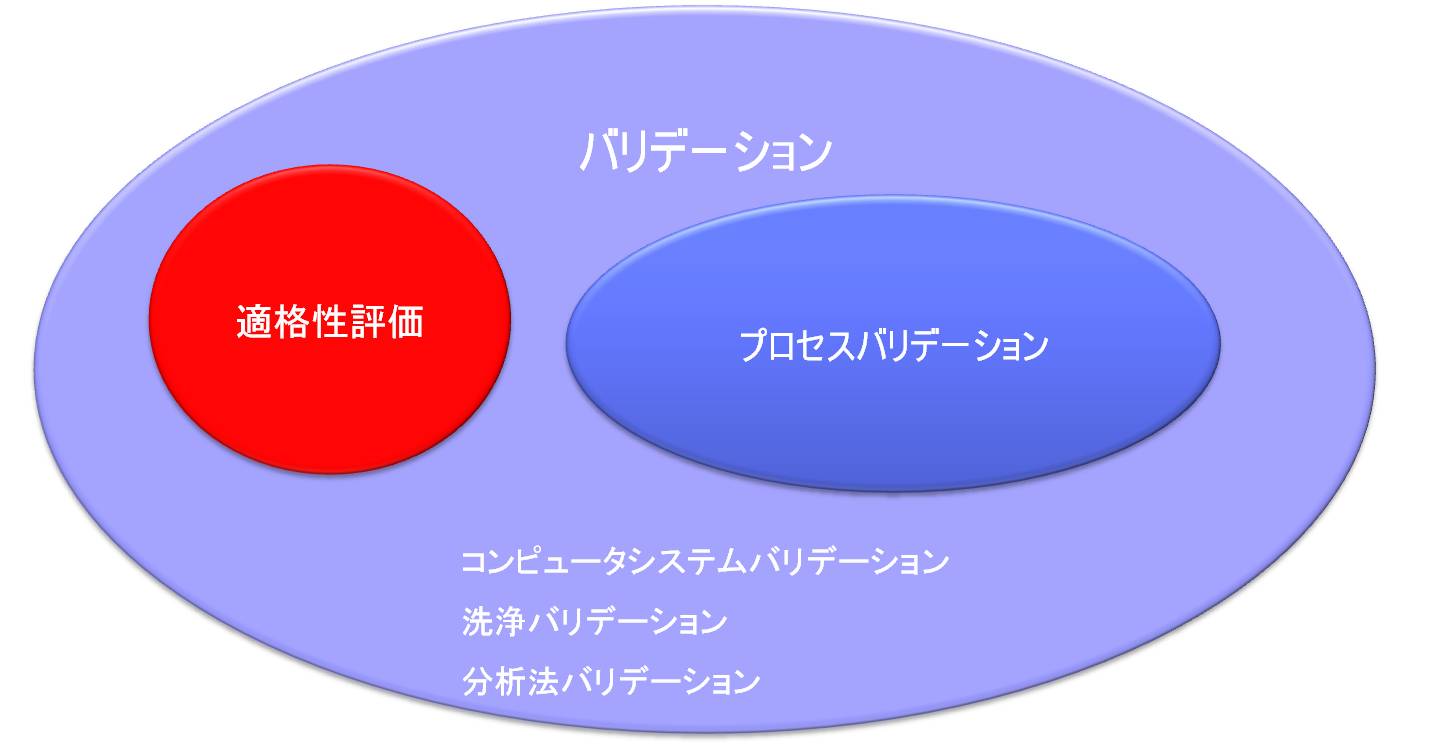
プロセスバリデーション活動
プロセスバリデーションは、バリデーション基準にはなぜか明確な定義が無く、原薬GMPのガイドライン中に記述がある。
設定パラメータ内で稼動する工程が、設定規格および品質特性に適合した製品を製造するために効果的かつ再現性よく機能できることに関する文書による確証のこと。
IQ:初期の適格性評価で、機器が必要とされ期待されたサービス内容を持つことの確認。(据付時適格性評価)
OQ:プロセスが許容される結果を生み、限界値(ワーストケース)を確立していることのデモンストレーション。(運転時適格性評価)
PQ:長期にわたるプロセスの安定性の確立。(性能適格性評価)
ソフトウェアのバリデーションについては、GMP省令、原薬のGMPガイドライン等に記載はないが、多くのプロセスがコンピュータ制御であることを認識すべきである。
GMPにおけるハードとソフト
GMPを語る上でははずせない要素・考え方として「ハード」と「ソフト」がある。
「GMPハード」と「GMPソフト」の両者によりGMPの目的を達成するのである。
GMPハードとは、設備のことであり、例えば以下のような要件である。
間違いを防ぐことのできる設備・環境の製造所であること
衛生的な設備・環境の製造所であること
高い品質を保ち続けることができる設備・環境の製造所であること
一方、GMPソフトとは、ルールのことであり、以下のような要件があげられる。
ルールを決めて文書化すること
ルールどおりに実施し、記録を作成すること
定期的に見直しを行い、改善をはかること
ここで、「GMPハード」と「GMPソフト」は、「ハードウェア」、「ソフトウェア」のことではないことに注意が必要である。「ハードウェア」、「ソフトウェア」はともに「GMPハード」である。
バリデーション(Validation)は、
- テスト(Test)
- 検証(Verification)
- 適格性評価(Qualification)
- 証明(Certificate)
- 監査(Audit)
- 照査(Review)
などを使って妥当性を検証するものである。
バリデーションの活動の中に適格性評価(クオリフィケーション)が含まれる。








