第6章 医療機器における査察
6.1 医療機器QMSの歴史とFDA規制の変遷
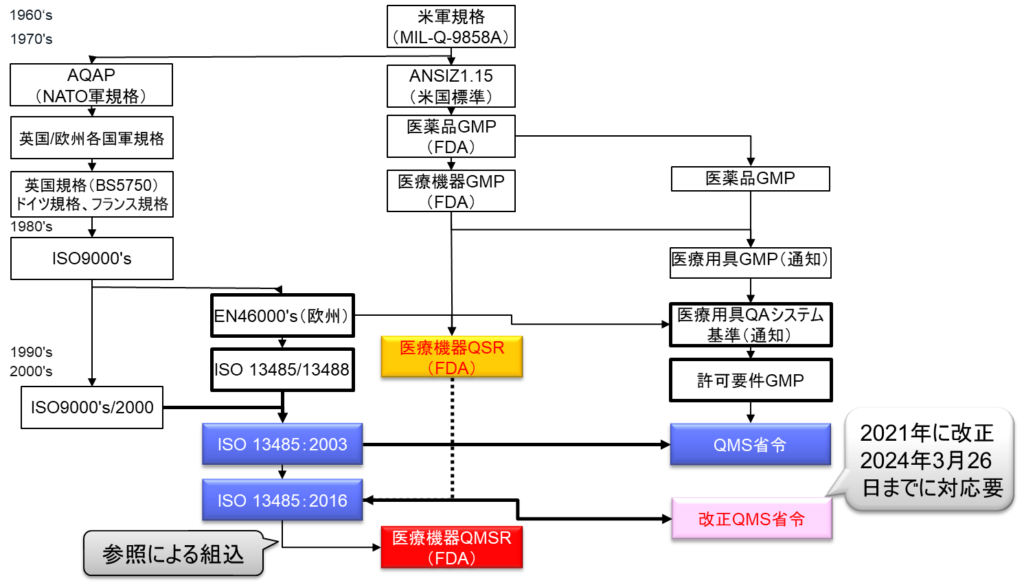
医療機器の品質管理システムは、1960年代初頭のサリドマイド事件を契機として大きく発展した。サリドマイドは当時、睡眠導入剤として販売されていたが、催奇形性という重大な問題を抱えていた。このサリドマイドを服用した妊婦から生まれた胎児に、四肢に異常が出るアザラシ症と呼ばれる奇形が発生し、ヨーロッパでは5,000例、日本でも350例以上が報告された。この悲惨な事件を受けて、医薬品の安全性に対する世論が沸き起こり、議会を動かすこととなった。議会はFDAに対してGMPの作成を命じ、その結果として1963年1月7日、FDAは世界で初めてGMPを発行した。これを受けて、医療機器に対するGMPも順次整備されていった。
1980年代に入り、FDAは医療機器特有の重要な課題に気づくこととなる。医薬品と異なり、医療機器は製造工程が適切であっても、設計そのものに問題があれば安全な製品とはならないという本質的な違いである。医薬品の場合、品質の問題の99%は製造所に起因するが、医療機器では製造工程の適切性に加えて、設計の妥当性が極めて重要となる。このため、製造(Manufacturing)に焦点を当てたGMPだけでは不十分であることが明らかとなった。
この認識に基づき、FDAは議会に働きかけてFD&C法(連邦食品・医薬品・化粧品法)の改正を実現し、設計から購買、さらにはサービスに至るまでの広範な領域での監査権限を獲得した。この権限拡大を受けて、1996年に医療機器GMPは大幅に改正され、医療機器QSR(Quality System Regulation)として生まれ変わった。この品質システム規則は翌1997年から施行され、その後長きにわたって医療機器の品質管理の基準として機能してきた。
一方、国際的な動きとして、ヨーロッパではISO 9001という品質マネジメントシステム規格が制定されていた。しかし、ISO 9001は顧客満足度の向上を主眼としており、医療機器に求められる安全性に関する規制要求を十分にカバーしていなかった。そこで、ISO 9001に安全要求事項を追加する形でISO 13485が策定された。2003年版のISO 13485は、初めての真のグローバルスタンダードとなり、日本の薬事法施行規則もこの規格との整合性を重視して運用されてきた。
2016年には、ISO 13485の大幅な改正が行われた。この背景には、旧版のISO 13485がFDAのQSRと比較して要求レベルが著しく低く、国際的な整合性を欠いているという課題があった。新しいISO 13485:2016は、FDAからの厳格な要求事項も反映する形で策定され、2021年には日本のQMS省令改正の際の参照規格として採用された。
しかし、ISO 13485:2016の発行により新たな課題も浮上した。アメリカ国内の企業は、米国内向けの申請ではQSRに、日本やヨーロッパ向けの申請ではISO 13485に従う必要があり、実質的に同等の要求事項に対して二重の対応を強いられる状況となった。この非効率的な状況を解消するため、FDAは2024年2月、ISO 13485:2016を完全に取り込んだ形で医療機器QMSRを最終規則として発行した。この改正により、医療機器の品質管理規制は日米欧で完全な整合化が図られることとなった。
6.2 Quality System Regulation (QSR)の基本的枠組み
Quality System Regulation(QSR)は、1996年10月7日にFDAによって公示された包括的な品質システム規則である。21 CFR Part 820として法制化されたこの規則は、人体用医療機器の設計から始まり、購買、製造、包装、ラベリング、保管、取り付け、そして付帯サービスに至るまでの全プロセスをカバーしている。
QSRの特徴的な点は、品質(Quality)を「安全性や性能を含めた使用適合性を満たすための機器の能力を支える特徴と特質の総体」と定義していることである。また、品質システムについては、「品質管理の実施に対する組織構造、責任、手順、プロセスおよび資源の総体」として明確に規定している。
QSRの一般規定では、各製造業者に対して、設計または製造される特定の医療機器に対して適切かつ規定の要件に適合する品質システムを確立し、維持することを求めている。この規定に定める要件は、最終機器が安全かつ有効であり、FD&C法(連邦食品・医薬品・化粧品法)の遵守を保証することを意図している。
なお、規則は最終機器の構成物や部品の製造業者には直接的には適用されないものの、このような製造業者に対してガイダンスとして規則の規定を用いることが推奨されている。QSRはサブパートAからOまでの15のセクションで構成され、それぞれが具体的な要求事項を規定している。総則から始まり、品質システム要求事項、設計管理、文書管理、購買管理、生産管理など、医療機器の品質管理に必要な全ての要素を網羅している。特に重要な点として、経営幹部の責任、品質監査、要員の要件などが詳細に規定されており、組織全体での品質マネジメントの実現を求めている。
規則の定義によれば、品質(Quality)とは安全性や性能を含めて使用適合性(Fitness-for-use)を満たすための機器の能力を支える特徴(Feature)と特質(Characteristic)の総体を指し、品質システム(QS)とは品質管理の実施に対する組織構造、責任、手順、プロセスおよび資源の総体を意味する。このように、QSRは単なる製造管理の枠を超えて、組織全体での品質マネジメントの実現を求めている。



6.3 QSIT (Quality System Inspection Technique)の概要と重要性
Quality System Inspection Technique(QSIT)は、FDA査察官のための包括的な査察マニュアルである。このマニュアルにより、査察官は一貫性のある効果的な査察を実施することが可能となる。近年では日本においてもQSIT査察が実施されており、その重要性は増している。
QSITの特徴的な点は、単なる製造管理や品質管理の確認に留まらず、品質保証システム全体を評価する点にある。特に、経営者によるマネジメントを中心に据え、設計から製造、品質保証、流通、据付に至るまでの全活動に対する適切な経営資源の配分と、それらの効果的な運用を重視している。
査察方法としては、トップダウン型のアプローチを採用している。これは、個々の事象から調査を開始するボトムアップ型とは異なり、まずシステム(文書)のレビューを行い、その後に実際の証拠(記録)の確認へと進むアプローチである。この方法により、組織の品質システムの全体像を効率的に把握することが可能となる。

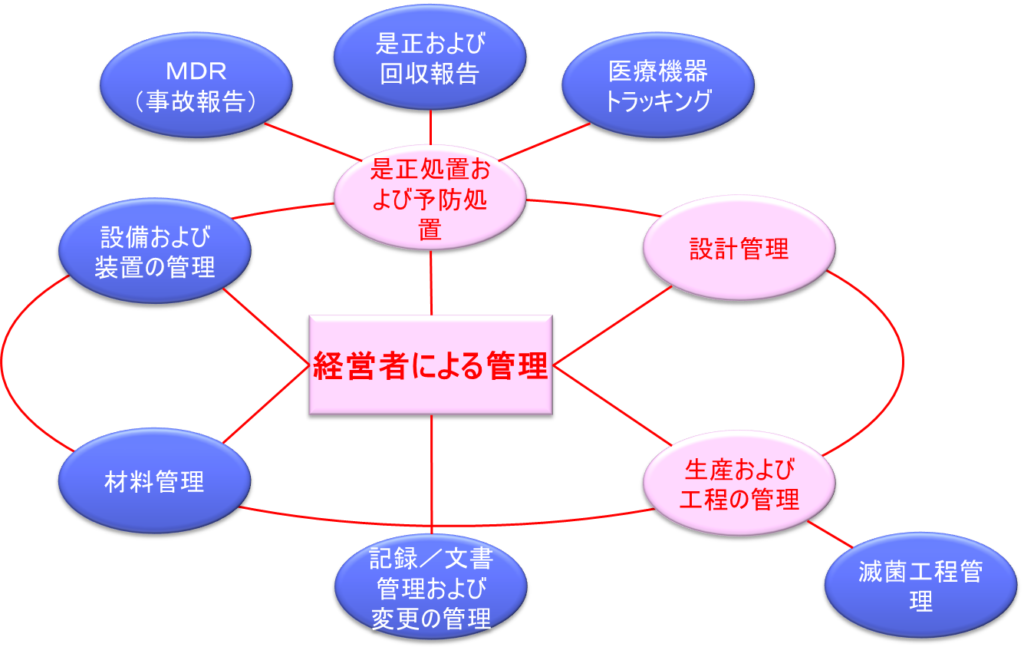
QSITガイドでは、品質システムの査察において重点的に確認すべき要素が体系的に整理されている。その中心には経営者による管理が位置づけられ、その周りを取り巻く形で複数の重要な管理要素が配置されている。
具体的には、是正および回収報告から始まり、是正処置および予防処置(CAPA)、医療機器トラッキング、MDR(事故報告)といった事後的な管理要素が含まれる。また、設備および装置の管理、設計管理、記録・文書管理および変更の管理、材料管理、生産および工程の管理、滅菌工程管理といった製造プロセスに直接関わる要素も重要な査察対象となっている。
これらの要素は互いに密接に関連しており、品質システム全体としての有効性を確保するために統合的に運用される必要がある。例えば、設計管理における変更は、記録・文書管理システムを通じて適切に管理され、必要に応じて生産および工程の管理にも反映されなければならない。同様に、MDRや是正処置の結果は、設計や製造プロセスの改善につながる可能性がある。
FDAの査察官は、これらの要素間の連携と統合の状況を詳細に評価し、組織の品質マネジメントシステムが実効的に機能しているかを判断する。特に、経営者の関与度合いは極めて重要視され、これらすべての要素に対する適切な資源配分と継続的な改善への取り組みが求められる。経営層には、品質システムの確立と維持に対する明確なコミットメントが求められ、それは具体的な行動や意思決定を通じて実証されなければならない。