
第5章: 製品開発と規制対応
5.1 製品開発プロセス
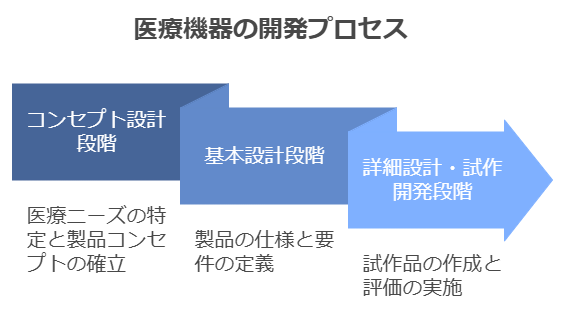
医療機器の製品開発は、一般的な製品開発とは異なり、開発の初期段階から規制要件を考慮する必要があります。まずコンセプト設計の段階では、医療ニーズの明確な特定から始める必要があります。この段階で重要なのは、臨床現場の具体的な課題を理解し、その解決に向けた製品コンセプトを確立することです。
基本設計段階では、製品の基本的な仕様を確定させます。この段階で特に重要なのは、設計インプットの明確化です。性能要件や安全性要件、使用環境要件など、製品に求められる要件を網羅的に特定する必要があります。また、各種規格・基準への適合性も、この段階から考慮に入れる必要があります。
詳細設計・試作開発の段階では、実際の製品仕様を確定させ、試作品の製作と評価を行います。医療機器の場合、設計検証には特に慎重なアプローチが求められます。安全性や有効性の評価に加えて、使用時のリスク評価など、多面的な検証が必要となります。
5.2 クラス分類と承認・認証・届出の選択
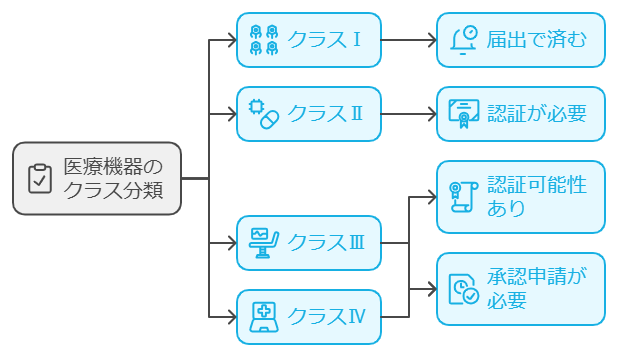
医療機器は、人体へのリスクの程度によって4つのクラスに分類されます。クラスⅠは不具合が生じた場合でも人体へのリスクが極めて低い機器、クラスⅡは比較的リスクの低い機器、クラスⅢは比較的リスクの高い機器、そしてクラスⅣは生命の危険に直結する可能性のある機器として分類されます。
このクラス分類に応じて、必要な手続きが異なってきます。クラスⅠ機器は届出で済みますが、クラスⅡの多くは認証が必要となり、クラスⅢ・Ⅳは原則として承認申請が必要です。ただし、クラスⅢ機器の一部は、認証基準が整備されている場合、認証で対応可能な場合もあります。
私の経験では、クラス分類の判断は開発の最初の段階で慎重に行う必要があります。クラス分類によって必要な対応が大きく異なるため、この判断が開発期間やコストに大きな影響を与えることになります。
5.3 QMS(品質マネジメントシステム)の構築
医療機器の製造販売には、適切な品質マネジメントシステム(QMS)の構築が不可欠です。QMSは単なる品質管理の仕組みではなく、設計開発から製造、市販後の監視まで、製品のライフサイクル全般をカバーする体系的なシステムです。
QMSの構築では、まず品質マニュアルの作成から始める必要があります。これは組織の品質方針や品質目標を明確にし、それを達成するための体制や手順を定めるものです。続いて、具体的な手順書類の整備を進めていきます。設計管理、製造管理、品質管理など、各プロセスに対応した手順書が必要となります。
特に重要なのは、設計管理プロセスの確立です。医療機器の設計開発は、各段階での検証と妥当性確認が求められます。設計インプットの確認から、設計アウトプットの検証、最終的な設計の妥当性確認まで、体系的なアプローチが必要です。
5.4 PMDAとの相談活用
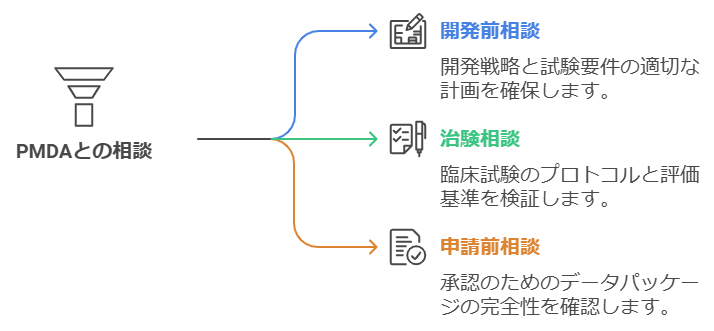
PMDAとの相談は、医療機器開発を効率的に進める上で極めて重要なツールとなります。相談の種類は開発段階によって異なり、開発前相談、治験相談、申請前相談など、様々な機会が用意されています。
開発前相談では、開発方針の確認や必要な非臨床試験の範囲、臨床試験の要否などについて、当局の見解を確認することができます。この段階での相談は、その後の開発計画を適切に立案する上で非常に有効です。
治験相談が必要な場合は、プロトコルの妥当性や評価項目の適切性など、臨床試験の計画に関する詳細な相談が可能です。また、申請前相談では、承認申請に必要なデータパッケージの充足性について確認することができます。
相談を効果的に活用するためには、十分な準備が必要です。相談資料には製品の概要や開発状況を簡潔かつ正確に記載し、具体的な相談事項を明確にする必要があります。また、相談結果は社内で適切に共有し、その後の開発活動に確実に反映させることが重要です。収載戦略について解説します。保険収載は、医療機器の市場性に大きな影響を与える重要な要素です。製品開発と並行して、早い段階から保険戦略を検討することが成功への近道となります。
本稿に関するご質問は下記まで。