医療機器に関する品質規則

医療機器の品質保証の特徴
単に製造と検査を管理するだけで医療機器の安全性を確保できるか?
- GMP的な決めたことの遵守と管理、不良品の発生防止だけでは機器の安全性を確保できるか?
- 医薬品と医療機器の特性の差(医薬品は化学物質、医療機器は?)
- 設計の重要性
- 使用方法(ユーザー要素)等予期し得ない安全性ファクターが大きい…リスクマネージメントの必要性
- 据付や付帯サービスが必要な機器もある
- 品質保証活動は製品提供の全分野に及ぶ
- 特にFDAは設計管理の重要性を認識
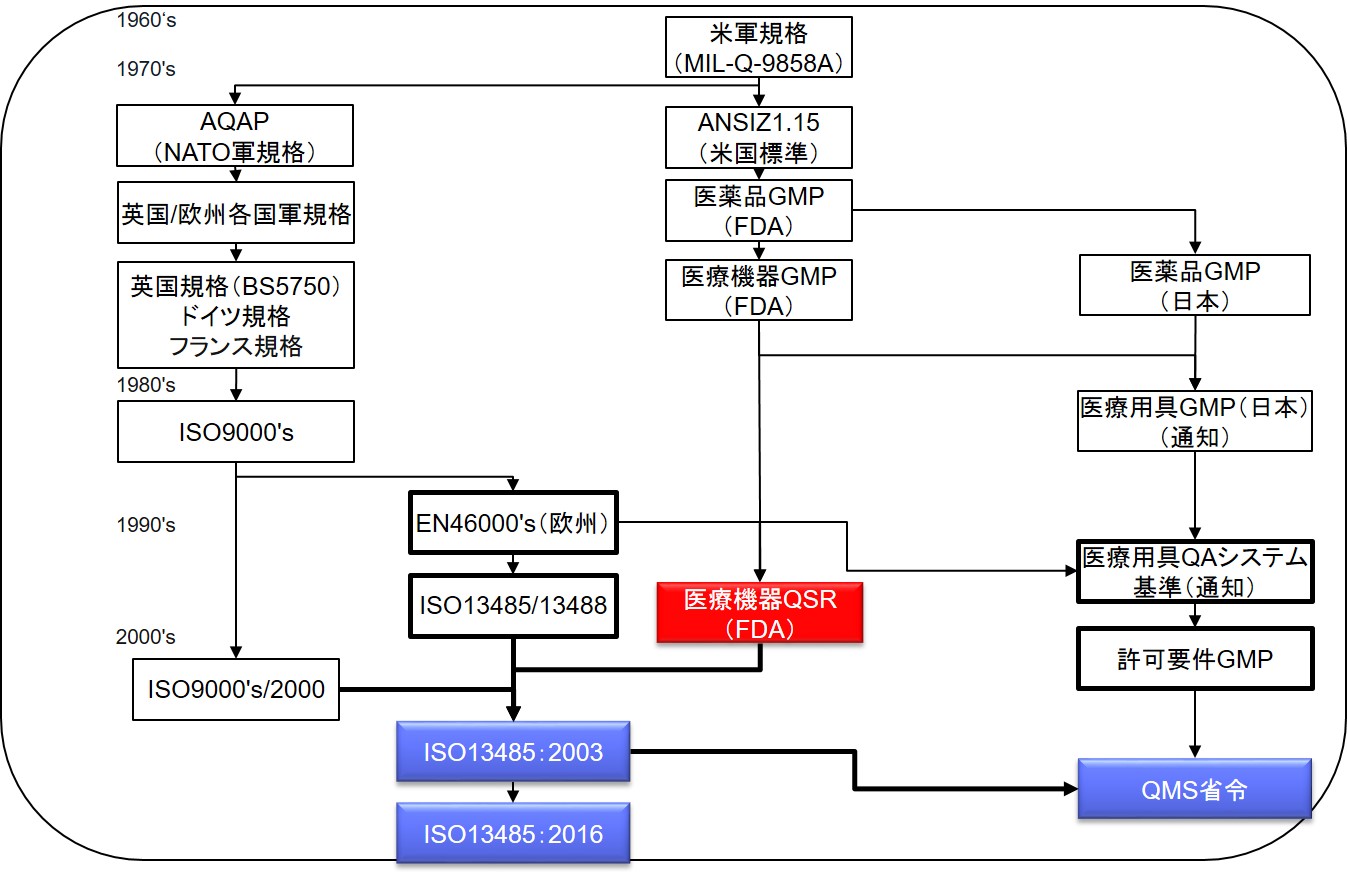
医療機器品質保証の国際調和
ISO9000‘sと追加要求事項規格ヨーロッパ規格のEN46000,s(1993)
GHTFによる5極調和化作業
1994日本GMPの改正(医療用具QAシステム基準)
一形の上ではISO9001+EN46001とほぼ整合がとられた
1995日本医療用具GMPの省令化
一QAシステム基準からのエッセンス(整合性には乏しい)
1997米国医療機器GMPの改正(医療機器QSR)
一設計管理の重要性を強調
新薬事法における医療機器QMS省令
品質システム規格の歴史
FDA近代化法(Food and Drug Administration Modernization Act: FDAMA)
1997年、FDAの行政改革を目的としてFDC法を改正する「FDA近代化法」がアメリカ議会を通過、同年11月21日、クリントン大統領の署名によって施行された。
この改正法は医薬品と医療機器に関する規制の強化と緩和が中心となっていて、多くの規則の改正や制定、あるいはガイダンスの作成を要求し、これまでにない広範な改革を求めるものであった。FDAは法律の発効後、3年以内に法律が要求する規則やガイダンスの仕事のほとんどを完了させた。
FDAMAには1993年度から発足した5年間の時限法の「処方せん薬ユーザーフィー法」を1997年度から5年間継続する規定(第二次ユーザーフィー法)も盛り込まれた(現在は2003年度から発足した第三次ユーザーフィー法が施行中)。
特に注目される改革には次のようなものがある。
- 小児研究の推進
- ファストトラック計画による承認審査の迅速化
- 医薬品の適応外使用(表示外適応)
- FDA規則の改善
- 医療機器の規制の強化と緩和
- 食品の包装材料の事前承認を廃止
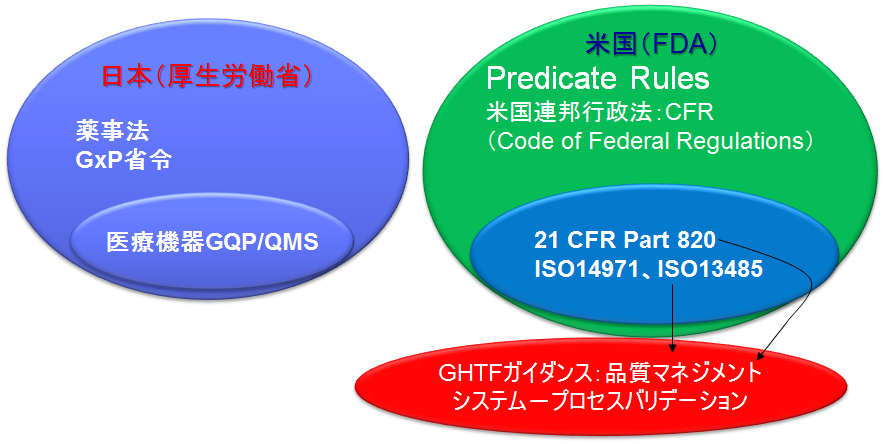
Code of Federal Regulation (CFR) Title 21
連邦規則集(連邦機関の現行規則を編集したもの)で、表題(Title)50のうち21にまとめられている。
Part 820は医療機器の品質システム規則
ISO13485はほぼ同様の内容を規定しており、日本ではJIS規格化されている。(JISは日本語として参照するのに適当)
Code of Federal Regulation (CFR) Title 21 Part 820
標題・・・Quality System Regulation: QSR(品質システム規則)
適用・・・人体用に意図された全ての医用完成機器の設計・購買・製造・包装・ラベリング・保管・取り付けおよび付帯サービス:医療機器の製造に関する基本的な要求事項
構成・・・サブパートAからOまでの15セクションに分かれている。
主な要件
- 品質システム要求事項
- 設計管理
- 文書管理
- 識別およびトレーサビリティ
- 生産および工程管理
- 是正処置および予防処置
品質システム規則:医療機器の製造に関する基準(医療機器GMP)
1996年10月7日、FDAは、1978年の医療機器GMP規則を改正して「品質システム」(QS)(Quality System)と呼ぶ新しい規則を公示した。「品質システム規則」(Quality System Regulation)(21 CFR 820)は、医療機器の設計、購入、製造、包装、表示、保管、設置およびサービスで用いられる手順とこれらに対して用いられる設備および管理に関する要件を含む。
規則の定義では、品質(Quality)は安全性や性能を含めて使用適合性(Fitness-for-use)を満たすため機器の能力を支える特徴(Feature)と特質(Characteristic)の総体をいい、QSは品質管理の実施に対する組織構造、責任、手順、プロセスおよび資源をいう。
1978年7月21日に公示されたGMP規則は医療機器の製造および品質管理に対する要件を定めているが、1990年に「医療機器安全法」(SMDA)が成立するまではほとんど変更がなかった。SMDAはGMP規則に設計管理(Design Control)の要件を盛り込むことや外国とのGMP相互認証協定への努力を求めた。FDAは、SMDAのもとにGMP規則を改正して新しく設計管理規定を加え、同時にGMP規則をできる限り国際基準(ISO9001)に含まれる品質システムの要件に合わせる方針をとった。
GMP規則改正の動きは、1990年4月の医療機器GMP諮問委員会の開催から始まった。この年から1995年にかけて、規則立案の事前公示(Advance Notice of Proposed Rulemaking)、一般からコメントを求める規則案の公示、GMP作業原案(Working Draft)の利用と原案に対するコメントを求める公示、GMP公聴会の開催、GMP諮問委員会の開催など、規則改正に向かって各種の手順が進むとともに、機器回収データの分析や国際品質基準などの評価が加わることによって、FDAはGMP改正を決断した。GMP規則は多くの異なるタイプの医療機器に適用しなければならないことから特定の機器に対する製造要件を詳しく規定することよりは、むしろすべての製造業者が守らなければならない大枠を定める方が合理的であると判断した。その趣旨にしたがってFDAは、製造業者の品質要件の達成に柔軟性を与えるため、規則案と作業班原案に変更を加えて最終GMP規則として「GMP : 品質システム規則」を公示した。
品質システム規則の一般規定は、次のようなことを定める。
- 各製造業者は設計または製造される特定の医療機器に対して適切な、そしてこの規定の要件に適合する品質システムを確立し、維持しなければならない。
- 規則に定める要件は最終機器が安全かつ有効であり、またFDC法の遵守を保証する意図をもつ。
- 規則は最終機器の構成物または部品の製造業者には適用されないが、このような製造業者に対してガイダンスとしてこの規則の規定を用いることを勧める。
また、外国の製造業者については、アメリカに輸入される医療機器の外国製造業者が施設へのFDA査察を拒否する場合、その施設で製造される機器は法的に不良品(Adulterated product)とみなされる。
規則は、一般規定(範囲、定義、品質システム)、品質システム要件(管理責任、品質査察、従業員)、設計管理、文書管理、購入管理、確認と追跡(確認、追跡)、生産および工程管理(生産および工程管理、検査・測定・検査器具、工程バリデーション)、容認作業(受領、工程中および最終機器の容認、容認の状態)、不適合製品、是正および予防措置、表示および包装管理(機器表示、機器包装)、取扱・保管・流通・設置(取扱、保管、流通、設置)、記録(一般要件、機器マスター記録、機器歴記録、苦情ファイル)、サービング、統計技術の各項目(Subpart)を含む。
General Principles of Software Validation
医療機器の設計、開発、製造に使用されるソフトウェアのバリデーションの原則を記述したもので、1987年に初版が発行され、2002年1月にファイナルガイダンスとして発行された。
本ガイダンスは、医療機器にかかわる業界およびFDAのスタッフに対するCSV関連の指針となっている。
規制の国際調和
グローバルに医療機器の規制要求事項を整合していくための組織が必要になり、官民合同の会議としてGHTF(The Global Harmonization Task Force)が結成。
GHTF SG 3(Study Group)=品質システムおよびプロセスバリデーションガイドライン=は、品質システムとしてISO13485:2003を採用するよう推奨しており、日本も薬事法改正の中でQMS省令を新たに発出し、この内容をISO13485:2003にあわせた経緯がある。
プロセスバリデーションは品質マネジメントシステム(QMS)要件の一部としている。
QMSには品質保証(QA)、設計管理、工程管理などが含まれる。
GHTFガイダンス
※21 CFR Part 820 (QMS)、ISO 13485をともに参照、網羅的に包含
バリデーション除外の場合:GHTFガイダンスでは、バリデーションが必要ない事象につき、ベリフィケーションを行うことでリスクやコストを低減でき得る、としている。
徹底したバリデーション実施の場合:バリデーションを緻密に行うことよりも製品の再設計をした方が、リスクやコストを低減できるかもしれない、という考え方がGHTFガイダンスにはある。
統計学的手法の活用:GHTFガイダンスでは、統計学的手法を特にOQ実施時に駆使し、製品スペックにおける管理範囲とアクションレベルを決定している。
ISO13485(医療機器の品質マネジメント)
ISO13485:2003の発行
ISO13485はISO9001と独立した規格であり、「Medical devices — Quality management systems — Requirements for regulatory purposes」として、世界各国で医療機器に関する規制目的で使用することを目的とした規格にモディファイされている。尚、日本国内では2005年10月1日にJIS Q 13485:2005としてJIS化された。
医療機器の品質管理(GQP)
医療機器製造販売業の許可を受けるためには、薬事法第12条の2第1号により、「品質管理の基準(Good Quality Practice:GQP)」に適合することが必要である。
GQPでは、主に「製品の市場への出荷の管理」、「適正な製造管理及び品質管理の確保(製造業者等の管理監督)」、「品質等に関する情報及び品質不良等の処理」など、品質管理業務を適切に実施するために必要なシステム(仕組み・ルール)の構築を要求している。
GQPでの要求事項は、製造販売業の許可の種類(第一種・第二種・第三種)によらず、全て共通となっている。
ISO14971(医療機器のリスクマネジメント)
EUでは1993年にMedical Devices Directive(MDD) 93/42/EECが施行され、機器をクラス分類により管理してきた。このクラス分類は主に機器の持つ危険の度合いによって振り分けられている。
MDDにおける危険回避は、すべての機器に対して基本要件の適合を義務づけることにより行われているが、この適合のためにリスク分析(prEN1441、1997年に正式版)が採用されてきた。
その後、EN1441に変えてリスクマネジメント(ENISO14971:2000)を整合規格に加え、今日に至っている。
現在もEU圏への医療機器の出荷にはリスクマネジメントが必須となっている。
QMSの管理浸透とリスクマネジメント
製造所の実務担当者と品質保証担当者は協力してQMS(品質マネジメントシステム)の管理浸透と適切なリスクマネジメントの実施を進めねばならない。
プロセスの理解とプロセスの継続的改善を達成するには設計およびプロセスバリデーションをしっかりおさえる必要がある。
